
Downloads
 Download
Download






This work is licensed under a Creative Commons Attribution 4.0 International License.
Review
Adrenergic Regulation of Cardiac Macrophages: Heterogeneity, Plasticity, and Therapeutic Potential
Wenjing Xiang †, Mianli Wang †, Hualong Yu †, Haocheng Lu *, and Ying Wang *
Department of Pharmacy, School of Medicine, South University of Science and Technology, Shenzhen 518055, China
† These authors contributed equally to this work.
* Correspondence: lhc@sustech.edu.cn (H.L.); wangy6@sustech.edu.cn (Y.W.)
Received: 8 June 2024; Revised: 10 July 2024; Accepted: 15 July 2024; Published: 25 October 2024
Abstract: Cardiac macrophages play a crucial role in the development and progression of cardiovascular diseases, including myocardial infarction, cardiac hypertrophy, and myocarditis. Macrophages are plastic cells that change their polarization states and functions in response to alterations in the surrounding environment. This process is deeply involved in various biological processes such as inflammation, tissue remodeling and repairing, exacerbating or mitigating the diseases progression. Thus, macrophages have emerged as potential therapeutic targets for multiple cardiac diseases. Upon sympathetic activation, adrenergic/ cyclic adenosine monophosphate (cAMP) signaling axis markedly modulates macrophages polarization and functions. It has been well-established that the intracellular cAMP is highly compartmentalized in cardiomyocytes. However, the spatiotemporal regulation of cAMP in cardiac macrophages and its implications in macrophage-driven cardiac diseases remain to be elucidated. In this review, we focus on the adrenergic/cAMP regulation of macrophage plasticity and function in the heart and discuss potentials and challenges of targeting the adrenergic/cAMP axis for cardiac diseases.
Keywords:
macrophage plasticity cAMP microdomains cardiac diseases phosphodiesterase1. Cardiac Macrophages Heterogeneity in Their Origins, Plasticity, and Functions
Cardiac macrophages are diverse cells originating from fetal monocytes or hematopoietic progenitors, crucially influencing heart health and disease [1,2]. They polarize into M1 (pro-inflammatory) or M2 (anti-inflammatory) phenotypes, adapting to environmental cues. In cardiac diseases like myocarditis and myocardial infarction, macrophages play pivotal roles in inflammation, regeneration, and repair [1]. They replace dead cells through efferocytosis and facilitate tissue repair by promoting angiogenesis and collagen deposition. The diverse functions and origins of macrophages significantly affect the heart’s response to injury and disease progression [1].
1.1. Cardiac Macrophages Have Diverse Origins
Cardiac macrophages comprise a diverse population of cells with various origins, which critically influence their function (Figure 1). Moreover, the macrophage population within the heart dynamically changes during diseases progression. For the past 40 years, it was hypothesized that all macrophages originated solely from monocytes. However, recent research supports the existence of heart-resident macrophages. These resident macrophages are categorized into two subsets: CCR2+ and CCR2- macrophages. CCR2- macrophages are derived from fetal monocyte progenitors and are maintained throughout the life independently of monocyte recruitment [3]. In contrast, CCR2+ macrophages, which originate from hematopoietic progenitors, are recruited to the heart during the early few weeks of life and are maintained through monocyte recruitment and subsequent proliferation. Following cardiac injury, resident macrophages are predominantly replaced by infiltrating monocytes and monocyte-derived macrophages [1].
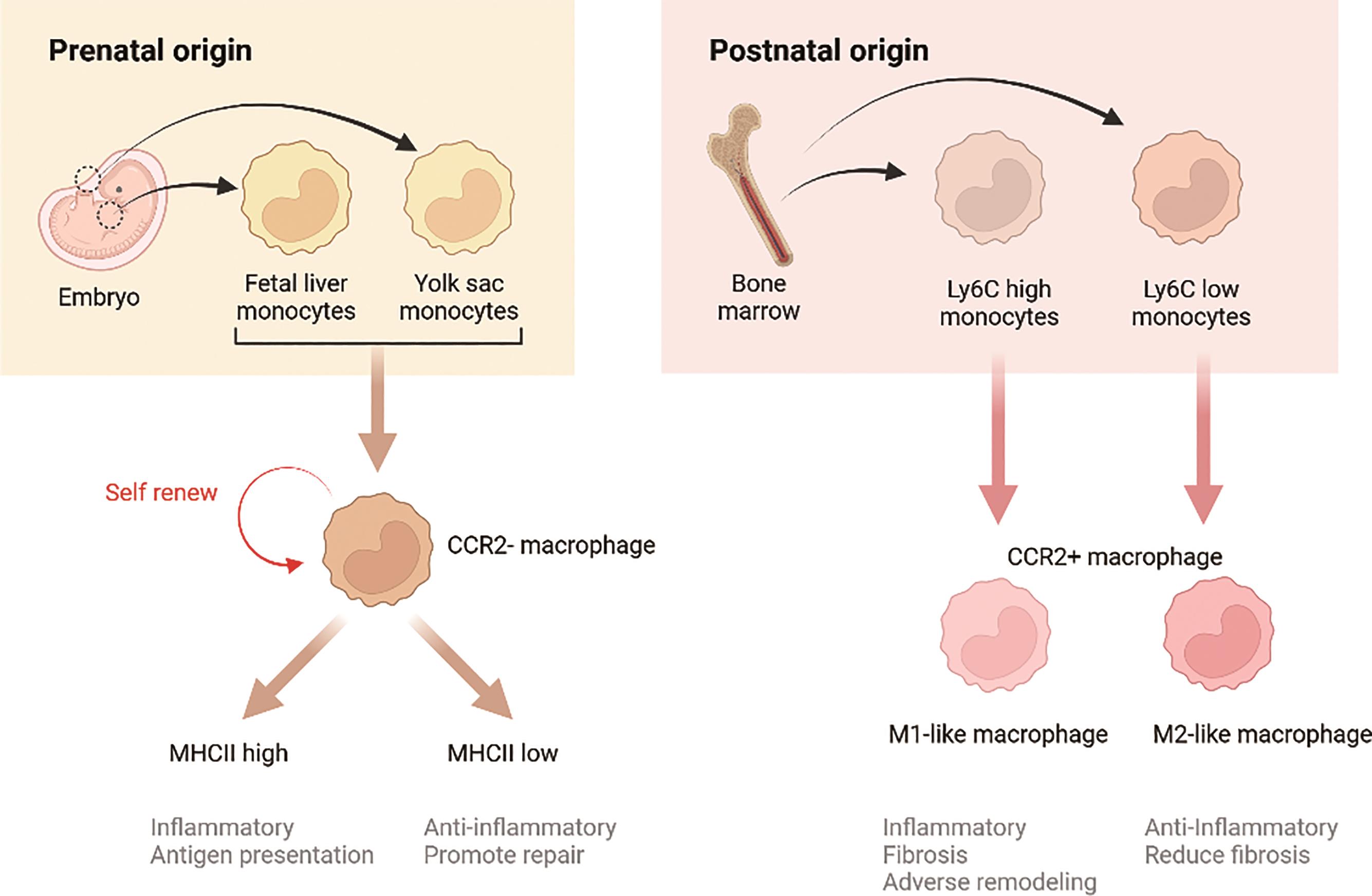
Figure 1. Origins and functions of cardiac macrophages.
1.2. Cardiac Macrophages Exhibit Remarkable Plasticity
Cardiac macrophages are remarkable plastic cells that can shift from one phenotype to another at distinct stages of various diseases [2] (Figure 1). Following exposure to micro-environment stimuli, macrophages polarize into specific phenotypes and functions. They are more commonly classified into two subsets: M1 and M2, based on their distinct phenotypes. Inflammatory M1 macrophages, also known as the classically activated macrophages, are pro-inflammatory, whereas alternatively activated M2 macrophages are anti-inflammatory. Dysregulation of M1/M2 polarization can lead to excessive inflammation and exacerbated cardiac injury. In a healthy heart, resident macrophages are typically M2-like, but they are lost during disease progression. M1 macrophages are typically induced by bacterial pathogens or Th1 inflammatory cytokines, while M2 are polarized by Th2 cytokines, including IL-4 and IL-13 [4]. Besides cytokines, a growing body of research supports that neurotransmitters, hormones, metabolites, and even mechanical stress can modulate the polarization, thus influencing disease development and progression. Cardiac macrophages can polarize and adjust their functional phenotypes in response to different stimuli in cardiac physiology and pathology, such as cytokines, chemokines, growth factors, pathogen-derived molecules etc. [5].
1.3. Cardiac Macrophages Are Deeply Involved in Cardiac Pathology and Repair
It is well recognized that macrophages play important roles in various cardiac diseases, being involved in all stages and modulate the heart biology (Figure 1). While macrophages are often categorized as pro-inflammatory M1 or pro-healing M2, recent studies have demonstrated a more complicated landscape of macrophages in heart.
1.3.1. Cardiac Inflammation
Myocarditis, characterized by inflammation of the myocardium in the presence of necrotic cells, is responsible for one in nine cases of heart failure (HF) and is a leading cause of heart transplantation, yet it still lacks targeted interventions. Myocarditis can be induced by parasites, viruses, or bacteria. In this condition, macrophages, along with neutrophils, constitute the main immune cell population.
Single-cell RNA sequencing (scRNA-Seq) has revealed the heterogeneity of macrophages in myocarditis [6,7]. In myocarditis induced by immune checkpoint inhibitors, the increased macrophage population mainly consists of CCR2+ monocyte-derived macrophage, which highly express Cxcl9, Cxcl10, Gbp2b, and Fcgr4 [8].
In autoimmune myocarditis, macrophages are characterized by the expression of Hif1a, with different macrophage clusters contributing to various disease stages [9]. In myocarditis induced by coxsackievirus B3 infection, most heart macrophages exhibit an M1-dominant functional phenotype, highly expressing nitric oxide synthase (NOS), interleukin-12 (IL-12), tumor necrosis factor-alpha (TNF-α), and CD16/32 [10,11]. The prolonged increased number of M1-like macrophage leads to chronic inflammation in heart [10]. The increased M1-like macrophage participated in antigen presentation, proinflammatory response, IL12 and IFNγ responses [9]. Notably, blocking CCR2 signaling prevents monocyte infiltration into the heart, thereby attenuating myocarditis and fibrosis [5].
1.3.2. Cardiac Regeneration
Macrophages are essential for heart regeneration in both neonatal and adult hearts. The neonatal heart can fully regenerate without forming scar after MI, but this regenerative capacity is disabled by depletion of neonatal macrophages [12]. Neonates with depleted macrophages fail to regenerate myocardium, leading to fibrosis, reduced cardiac function, and impaired angiogenesis. Interestingly, in relative to non-regenerative macrophages, regenerative macrophages exhibit a unique polarization profile, expressing several soluble factors, angiogenesis genes, and oxidative stress-related genes. Regenerative macrophages are predominantly mediated by cardiac-resident macrophages. This is evidenced by experiments where transferring monocytes from adult mice spleens to neonatal mice hearts following MI injury led to fibrosis rather than regeneration, indicating that infiltrating monocytes minimally contribute to the cardiac regeneration. Furthermore, depletion of cardiac-resident macrophages by deleting colony stimulating factor 1 receptor (Csf1r) homologue impairs cardiac regeneration without affecting monocyte-derived macrophages [13]. Inhibiting monocyte infiltration to the heart by CCL2-CCR2 signaling blockade increases CCR2- resident macrophages proliferation.
1.3.3. Efferocytosis of Dead Cardiomyocyte
Efferocytosis is essential for maintenance of heart homeostasis through the clearance of apoptotic cells by non-professional phagocytes. Myocardial infarction causes cardiomyocyte death and then triggers immune responses and macrophages-mediated efferocytosis [14,15]. After MI, circulating monocytes are recruited to the injured heart where resident macrophages are replaced by CCR2+ monocyte-derived macrophages [16]. These monocyte-derived macrophages can be further divided into three classes, two pro-inflammatory populations defined as Isg15hi and MHCII+Il1b+, alongside non-inflammatory Trem2hi cells [17]. Pro-inflammatory macrophages play a crucial role in scavenging damaged or dead cells through efferocytosis, which is critical for resolving inflammation after MI [12]. During efferocytosis, macrophage membrane receptors including purinergic receptor P2Y2, G-protein-coupled receptors G2A and S1P1-5 sense apoptotic cells [13]. Then, macrophages recognize “eat-me” signals on the surface of apoptotic cells by MerTK, CD36, integrins αvβ3 and αvβ5, TIM-1,4 [T-cell membrane protein (Tim) family], BAI1 (brain angiogenesis inhibitor 1), and stabilin-2 [18]. Apoptotic bodies are then engulfed by macrophages and degraded in lysosomes [19]. Resident macrophages express legumain, which promotes the clearance of apoptotic cardiomyocytes and improves cardiac repair [16]. Interestingly, efferocytosis also induces adaptive responses in macrophages, including triggering VEGFC secretion, inducing proliferation [17], and promoting metabolic adaptation [14,20]. These adaptative responses help maintain macrophage fitness and tissue homeostasis.
1.3.4. Cardiac Repairing
Macrophages are crucial for tissue repair. The heart gradually loses the regeneration capacity after birth and the macrophage pattern also differs in neonatal and adult heart. Neonatal mouse heart contains one macrophage (MHC-IIhigh CCR2+) and one monocyte population (MHC-IIlow CCR2+). The MHC-IIhigh CCR2+ macrophage expanded after neonatal heart injury without recruiting CCR2+ monocytes from circulation [21]. Instead, the injured adult heart mainly recruited CCR2+ monocytes and CCR2- resident macrophages are depleted after injury [22]. In the injured heart, macrophages promote angiogenesis by stimulating the proliferation of endothelial cells and smooth muscle cells [23]. Since adult cardiomyocytes exhibit minimal regeneration ability, the infarcted sites are often repaired by fibrosis. Macrophages secrete TGF-β and fibroblast growth factors to activate fibroblast and myofibroblast, leading to collagen deposition [24]. Additionally, macrophages can also directly synthesize collagen and proteoglycans, participating actively in tissue repair [25,26]. In the neonatal heart, macrophages also promote cardiomyocyte dedifferentiation and proliferation, aiding in heart regeneration [27].
1.3.5. Fibrosis and Tissue Remodeling
Inflammation is a key mechanism by which the heart responds to injury and undergoes adaptive remodeling. Cardiac dysfunctions associated with obesity or diabetes, such as diabetic cardiomyopathy and obesity cardiomyopathy, are characterized by excessive M1 macrophages polarization and activation.
After heart injury, it is critical to replace the damaged area with scar tissue to preserve ventricular integrity and heart function. However, extensive scar formation can lead to adverse cardiac remodeling. The extent of scar formation is determined by the balance between extracellular matrix production and degradation [1]. Cardiac macrophages can induce the endothelial-to-mesenchymal transition (EMT) of endothelial cells, via MMP14 and TGF-β [9]. The EMT process results in a fibroblast-like phenotype, contributing to cardiac fibrosis. Inflammatory cytokines from macrophages, including IL-1α, IL-1β, IL-6 and IL-8 activate fibroblasts, increasing the secretion of IL-6 and TGF-β from both macrophages and fibroblasts. This activation leads to increased proliferation and collagen production by fibroblasts [28,29]. Interestingly, the M2b macrophage subset suppresses the proliferation and migration of fibroblast, alleviating cardiac fibrosis [30]. This demonstrates that different subsets of macrophages may play distinct roles in cardiac fibrosis.
2. Adrenergic-cAMP Signal Modulates Macrophages
Accumulating evidence indicates that the sympathetic nervous system plays a pivotal role in modulating immune system, particularly macrophages [31]. The sympathetic nervous system critically influences macrophage function through adrenergic receptors and cAMP signaling, modulated by phosphodiesterases (PDEs) [32,33]. Upon physiological and pathological stress conditions, catecholamines such as norepinephrine or epinephrine are released and bind to adrenergic receptors on macrophages, increasing intracellular cAMP levels. PDEs break down cAMP, controlling the magnitude and duration of the signal [34,35]. This adrenergic-cAMP-PDE signaling system fine-tunes macrophage functions and presents potential therapeutic intervention targets [33‒35]. However, conflicting observations exist regarding the individual modulation of macrophages by activating adrenergic receptors, cAMP, and PDEs, indicating a need for further research to integrate and understand these mechanisms fully.
2.1. Adrenergic Receptors (ARs) in Macrophages
The activation of adrenergic receptors in macrophages exhibits bidirectional and multifaceted roles. Elevated sympathetic tone can promote M1 macrophages in the spleen through direct innervation, polarizing macrophages toward an inflammatory M1 phenotype and enhancing monocyte production. Similarly, in vivo stimulation of adrenergic receptors by isoproterenol activates the NLRP3 inflammasome, leading to pro-inflammatory macrophage infiltration in the mouse heart [36]. After MI, elevated circulating catecholamine levels activate macrophages and promote post-MI arrhythmias [37]. Conversely, AR activation by norepinephrine is well-established in promoting anti-inflammatory and tissue-reparative phenotype in macrophages. Macrophages express both α (α1, α2 and α3) and β-ARs (β1, β2 and β3). Norepinephrine has been found to increase macrophage fatty acid uptake and triglyceride storage by activating β2-AR, which is similar to the features of macrophages isolated from mouse hearts after myocardial injury [38]. Deleting β2-AR in macrophages inhibits their polarization towards M2 [39], thereby impairing β2-AR-mediated cardiac repair and survival post-MI. This deletion disrupts the recruitment of monocyte-derived macrophages to the injured heart to repair, causing heart rupture. In muscularis macrophages, β2-AR activation protects against infection-induced cell death by enhancing arginase 1-polyamine-depent M2 polarization. Activating β3-AR promotes tissue macrophage accumulation and lipolysis in adipose tissue, though less is known about the macrophages-expressed β3-AR in cardiac diseases progression. Furthermore, β2-AR and β3-AR activation synergistically promote M2 macrophages infiltration. Unlike β2-AR or β3-AR, the role of β1-AR in macrophages and its implication in cardiac diseases are less investigated. Moreover, despite the roles of β2-AR or β3-AR in promoting anti-inflammatory and reparative M2 macrophages, clinical trials demonstrate that β-AR blockers such as metoprolol, carvedilol and bisoprolol alleviate cardiac inflammation and macrophages infiltration [40‒43]. Notably, there are huge differences among individual β-blockers’ effects on macrophages and inflammatory responses. For example, carvedilol reduces LPS-induced Toll-Like Receptor 2 expression and inflammation in macrophages cell lines (RAW 264.7 cells) [44]. In vivo administration of carvedilol reduces macrophages accumulation and M1-macrophages polarization. Both intravenous administration of carvedilol and metoprolol significantly reduce macrophage infiltration in MI pig hearts, with carvedilol exhibiting stronger effects than metoprolol [45]. This aligns with the clinical meta-analyses showing that carvedilol has more beneficial effects on patients’ LV remodeling compared to metoprolol [46]. Interestingly, metoprolol, but not atenolol or propranolol, attenuates cardiac inflammation in mouse ischemia-reperfusion model. In silico predictions indicate this is due to the unique conformational changes of intracellular adrenergic receptors induced by metoprolol, which are not observed within atenolol or propranolol [41].
In addition to βARs, α-ARs have also been identified as key regulators of macrophages. For instance, activation of α1-AR in macrophages promotes M2 macrophages polarization, thereby enhancing cardiomyocyte proliferation and angiogenesis [47]. On the other hand, antagonism of α2A-AR attenuates LPS-induced M1 macrophages polarization by down-regulating MAPK signaling [2]. The molecular mechanisms underlying the significant different impacts of adrenergic agonism or antagonism remain to be elucidated (Figure 2). Understanding these mechanisms is crucial to provide insights into therapeutic strategies targeting adrenergic receptors to modulate macrophage functions and improve outcomes in cardiac diseases.
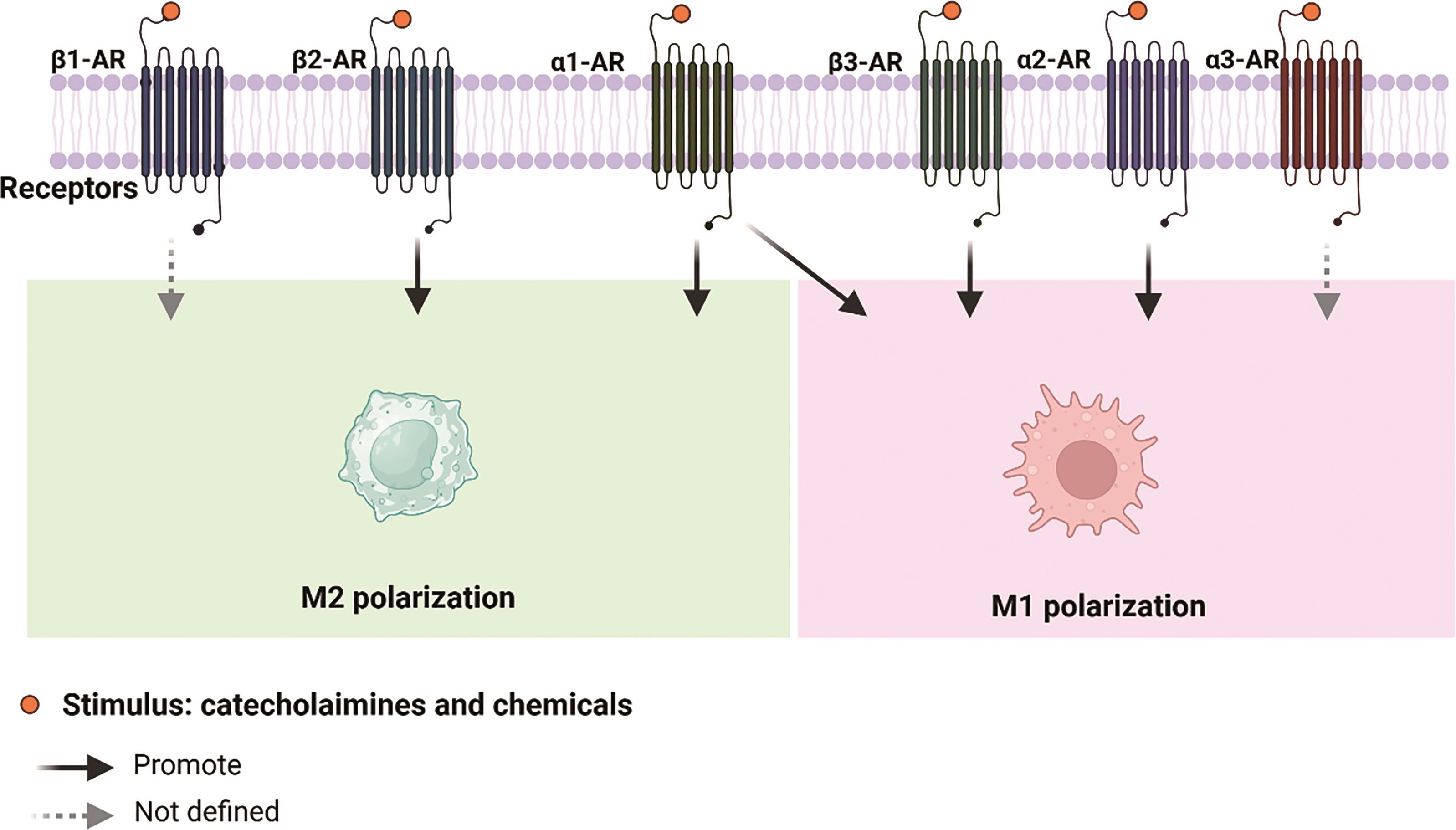
Figure 2. Individual adrenergic receptor activation impacts macrophages plasticity.
2.2. cAMP-PKA
Elevating cAMP levels significantly regulates of macrophages polarization and functions, including phagocytosis, metabolic reprogramming and efferocytosis [48,49]. Increased cAMP inhibits polarization towards the pro-inflammatory M1 phenotype, reducing the expression of M1 markers such as inducible cyclooxygenase (COX-2) and inducible NOS (iNOS). Elevated cAMP transforms M1 macrophages into resolution-phase macrophages that promote the resolution of systemic inflammation, implicating cAMP in resolving cardiac inflammation [48]. In the presence of IFN-γ/LPS, db-cAMP (a cAMP mimetic) can switch M1 macrophages towards an M2-like phenotype. Similarly, enhancing cAMP amplifies the IL-4-induced M2 phenotype and promotes macrophages to express engulfment molecules and efferocytosis of apoptotic neutrophils [48]. Additionally, cAMP is an important regulator of monocytic differentiation into macrophages and their functions. In the presence of cAMP, GM-CSF-induced monocyte differentiation is altered, promoting the expression of wound-healing cytokines, such as IL-10 and IL-6. Furthermore, enhancing cAMP levels with forskolin prevents macrophages from expressing several surface markers, including CD14, the coreceptor for LPS. Enhancing cAMP production by dibutyryl cAMP or stimulating adenyl cyclase by forskolin, prevents lipopolysaccharide-induced TNF-α production in macrophages.
As the major effector of the second message cAMP, PKA critically influences macrophages phenotypes. Activation of PKA promotes M2 polarization via CREB-mediated aerobic glycolysis [50]. Further, PKA has been demonstrated to negatively regulate pyroptosis and inflammasome activation. PKA-dependent phosphorylation of either caspase-11 or NLRP3 prevents pyroptosis or inflammasome activation. These effects are largely abolished by PKA inhibitor, highlighting the central role of cAMP-PKA in modulating macrophage responses [51,52].
2.3. Phosphodiesterase
Targeting phosphodiesterases (PDEs) with small-molecule compounds for inflammatory disorders has been an area of active research interest for many years [32]. Despite the enriched knowledge of PDE expression and regulation in immune cells such as macrophages, less is known about the role of PDEs in cardiac inflammatory responses [53]. Macrophages express all PDE1-11 families, with significant changes in PDE profiles observed during the differentiation of human monocytes into macrophages [54]. Each PDE subfamily has its unique substrate selectivity and pharmacology [32,35]. PDE4, 7, and 8 are exclusively involved in cAMP hydrolysis, whereas PDE1,2,3, 10 and 11 can hydrolyze both cAMP and cGMP. Among them, PDE3 is the principal PDE isoenzyme expressed in pro-inflammatory cells, including macrophages [55]. During the in vitro differentiation, PDE1 and PDE3 activity increases, while the major cAMP-hydrolyzing enzyme, PDE4, rapidly declines. Correspondingly, inhibiting PDE1 by vinpocetine reduces macrophage secretion of inflammatory proteins [56]. Inhibition of PDE3 by cilostazol reduces macrophages recruited to injury tissues. Polarization of M1 macrophages by LPS is associated with significant increases in PDE4B and PDE10A [57]. LPS induces a sustained expression of PDE10A and a transient expression of PDE4B in macrophages. Inhibiting PDE4B blocks TNF-α expression, whereas inhibiting PDE10A abolishes MCP-1 expression upon LPS stimulation, highlighting the distinct roles of PDEs and PDE-modulated cAMP in regulating macrophage phenotypes. While neither PDE3 nor PDE4 inhibition alone reduces LPS-induced TNF release in monocyte-derived macrophages, their combined inhibition results in a 40–50% reduction of LPS-induced TNF-α [54]. Distinguished from the PDEs listed above, the role of PDE2 and PDE11 in macrophages remains unknown. Overall, targeting PDEs presents a promising strategy for modulating macrophage function and inflammatory responses in cardiac diseases. Understanding the specific roles of different PDEs in macrophage biology could lead to more effective and targeted therapies for inflammatory and cardiac conditions.
3. New Directions and Challenges Targeting Adrenergic Regulation of Macrophages for Cardiac Diseases
3.1. Understanding the Heterogeneity of Macrophages in the Remodeled Heart
Macrophages play a crucial and diverse role in the diseased heart, contributing to processes such as inflammation, tissue repair, fibrosis, and irregular electrophysiology. Understanding their heterogeneity and functional diversity is essential for developing targeted therapies that selectively modulate specific macrophages populations, potentially improving treatment outcomes for cardiac diseases. However, fully unraveling macrophage heterogeneity and its molecular mechanisms in cardiac diseases poses significant challenges. First, cardiac macrophages derive from different origins exhibit dynamic phenotypic changes in response to the local micro-environment, making them difficult to dissect and profile. Second, macrophages interact with numerous cell types in the heart through direct coupling or by secreting a wide series of cytokines, chemokines, and growth factors that affect cardiac function. Understanding these complex signaling networks require comprehensive and integrative proteomic and transcriptomic analyses. Fortunately, advancements in single-cell sequencing techniques now enable the detailed profiling of macrophages heterogeneity through single-cell transcriptional, metabolic or proteomic sequencing. These techniques can unmask the diverse profiles of macrophages at unprecedented resolution. Additionally, sophisticated lineage-tracing techniques allow the identification of macrophages derived from distinct origins in both physiological and pathological conditions. Characterizing the heterogeneous macrophages in the diseased heart requires integrating insights from single-cell technologies and advanced linage-tracing techniques. Revealing the specific roles and interactions of various macrophage subpopulations may ultimately promote the development of more precise and effective therapeutic strategies for cardiac diseases.
3.2. Defining Compartmentalization of AR/cAMP in Macrophages
Over the last two decades, it has become well-acknowledged that pools of cAMP are discretely located to distinct subcellular compartments, where they transduce signals to different downstream targets and cell functions. For instance, the cAMP microdomain at endoplasmic reticulum or sarcoplasmic reticulum modulate calcium uptake through PKA-dependent phosphorylation of phospholamban, the inhibitory protein for the calcium pump. The ER/SR-localized cAMP microdomain does not affect PKA activity at PM and thus does not impact activities of local ion channels, including L-type calcium channels and RyR. Recent studies suggest that nuclear-localized cAMP is a key regulator of histone transcription and cell proliferation [58]. Various cAMP microdomains in cardiomyocytes and neurons have been unmasked with the development of subcellular-anchored Förster resonance energy transfer (FRET)-based biosensors of cAMP and PKA [59‒62]. Utilizing FRET biosensors, insight into the spatiotemporal regulation of cAMP-PKA dynamics is rapidly growing. However, the subcellular characterization of cAMP in macrophages is still lacking. Furthermore, very little is known about the impact of individual cAMP microdomains on macrophages functions in healthy and diseased hearts. The compartmentalized cAMP microdomains in different macrophage subpopulations may reconcile the diverse roles of NE stimulation and PDEs inhibition in regulating macrophage functions. Understanding these subcellular dynamics and their specific impacts on macrophage behavior could provide valuable insights into the development of targeted therapies for cardiac diseases.
3.3. Leveraging PDE Isoform-Selective Inhibition to Target Distinct AR/cAMP Compartments
Despite the encouraging preliminary observations of anti-inflammatory results, several PDE inhibitors have failed in clinical trials for cardiovascular diseases treatment. For instance, clinical studies with PDE4 inhibitor as an anti-inflammatory agent have only met with limited success [32]. Several factors must be considered for the development of therapeutic targeting PDEs in macrophages for potential cardioprotective effects. First, many PDE isoforms have their unique subcellular expression patterns or are recruited to distinct compartments to regulate specific cell functions. A detailed understanding of the distribution, regulation, and function of individual PDE isoforms in macrophages is still lacking. Each PDE isoform may play different roles depending on its localization and interaction with other cellular components. Second, chemical inhibitors globally interrupt a subfamily of PDEs without specificity to certain isoforms, which can lead to limited efficacy and unwanted side effects. This lack of specificity is a significant hurdle to cause off-target effects and compromise the therapeutic potential of these inhibitors. To address these challenges, future drug development could focus on selective peptides or small inhibitory RNA for individual PDE isoform and even splice variants [63]. Such precision could enhance the effectiveness of treatments by modulating the activity of specific PDE isoforms involved in pathological processes, while minimizing adverse effects.
4. Conclusions
Adrenergic signaling plays a multifaceted role in regulating macrophages in various cardiac diseases. Utilizing advanced single-cell sequencing, subcellular-anchored biosensors, and lineage-tracing techniques to dissect the compartmentalized adrenergic-cAMP-PDE signaling and their implication in the modulation of macrophages heterogeneity and plasticity may provide innovative therapeutic targets for cardiac diseases.
Author Contributions: Y.W. and H. L. conceived the idea; M.W., H.Y., W.X. H.L. and Y.W.; drafted the manuscript; Y.W., M.W. and H.Y. critically edited the manuscript; All authors have read and agreed to the published version of the manuscript.
Funding: This work was supported by National Natural Science Foundation of China (K231141004, K23411108 and 82070406) and Guangdong Basic and Applied Basic Research Foundation (2024A1515010543).
Institutional Review Board Statement: Not applicable.
Informed Consent Statement: Not applicable.
Data Availability Statement: Not applicable.
Acknowledgments: We acknowledge Haibo Ni and Chaoshan Han for his/her generous contributions in language editing and intellectual refinement of this manuscript.
Conflicts of Interest: The authors declare no conflict of interest.

References
- Yap, J.; Irei, J.; Lozano-Gerona, J.; et al. Macrophages in Cardiac Remodelling after Myocardial Infarction. Nat. Rev. Cardiol. 2023, 20, 373–385.
- Sica, A.; Mantovani, A. Macrophage Plasticity and Polarization: In Vivo Veritas. J. Clin. Investig. 2012, 122, 787–795.
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; et al. Embryonic and Adult-Derived Resident Cardiac Macrophages Are Maintained through Distinct Mechanisms at Steady State and during Inflammation. Immunity 2014, 40, 91–104.
- Chen, Y.; Hu, M.; Wang, L.; et al. Macrophage M1/M2 Polarization. Eur. J. Pharmacol. 2020, 877, 173090.
- Vadevoo, S.M.P.; Gunassekaran, G.R.; Lee, C.; et al. The Macrophage Odorant Receptor Olfr78 Mediates the Lactate-Induced M2 Phenotype of Tumor-Associated Macrophages. Proc. Natl. Acad. Sci. USA 2021, 118, e2102434118.
- Law, Y.M.; Lal, A.K.; Chen, S.; et al. Diagnosis and Management of Myocarditis in Children: A Scientific Statement from the American Heart Association. Circulation 2021, 144, e123–e135.
- Hua, X.; Hu, G.; Hu, Q.; et al. Single-Cell RNA Sequencing to Dissect the Immunological Network of Autoimmune Myocarditis. Circulation 2020, 142, 384–400.
- Ma, P.; Liu, J.; Qin, J.; et al. Expansion of Pathogenic Cardiac Macrophages in Immune Checkpoint Inhibitor Myocarditis. Circulation 2024, 149, 48–66.
- Lafuse, W.P.; Wozniak, D.J.; Rajaram, M.V.S. Role of Cardiac Macrophages on Cardiac Inflammation, Fibrosis and Tissue Repair. Cells 2020, 10, 51.
- Dong, J.; Lu, J.; Cen, Z.; et al. Cardiac Macrophages Undergo Dynamic Changes after Coxsackievirus B3 Infection and Promote the Progression of Myocarditis. J. Med. Virol. 2023, 95, e29004.
- Li, K.; Xu, W.; Guo, Q.; et al. Differential Macrophage Polarization in Male and Female BALB/c Mice Infected with Coxsackievirus B3 Defines Susceptibility to Viral Myocarditis. Circ. Res. 2009, 105, 353–364.
- Peet, C.; Ivetic, A.; Bromage, D.I.; et al. Cardiac Monocytes and Macrophages after Myocardial Infarction. Cardiovasc. Res. 2020, 116, 1101–1112.
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; et al. Nucleotides Released by Apoptotic Cells Act as a Find-Me Signal to Promote Phagocytic Clearance. Nature 2009, 461, 282–286.
- Gerlach, B.D.; Ampomah, P.B.; Yurdagul, A.; et al. Efferocytosis Induces Macrophage Proliferation to Help Resolve Tissue Injury. Cell Metab. 2021, 33, 2445–2463.e8.
- Doran, A.C.; Yurdagul, A.; Tabas, I. Efferocytosis in Health and Disease. Nat. Rev. Immunol. 2020, 20, 254–267.
- Bajpai, G.; Bredemeyer, A.; Li, W.; et al. Tissue Resident CCR2- and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ. Res. 2019, 124, 263–278.
- Rizzo, G.; Gropper, J.; Piollet, M.; et al. Dynamics of Monocyte-Derived Macrophage Diversity in Experimental Myocardial Infarction. Cardiovasc. Res. 2023, 119, 772–785.
- Elliott, M.R.; Ravichandran, K.S. The Dynamics of Apoptotic Cell Clearance. Dev. Cell 2016, 38, 147–160.
- Kinchen, J.M.; Ravichandran, K.S. Phagosome Maturation: Going through the Acid Test. Nat. Rev. Mol. Cell Biol. 2008, 9, 781–795.
- Glinton, K.E.; Ma, W.; Lantz, C.; et al. Macrophage-Produced VEGFC Is Induced by Efferocytosis to Ameliorate Cardiac Injury and Inflammation. J. Clin. Investig. 2022, 132, e140685.
- Lavine, K.J.; Epelman, S.; Uchida, K.; et al. Distinct Macrophage Lineages Contribute to Disparate Patterns of Cardiac Recovery and Remodeling in the Neonatal and Adult Heart. Proc. Natl. Acad. Sci. USA 2014, 111, 16029–16034.
- Dick, S.A.; Zaman, R.; Epelman, S. Using High-Dimensional Approaches to Probe Monocytes and Macrophages in Cardiovascular Disease. Front. Immunol. 2019, 10, 2146.
- Fantin, A.; Vieira, J.M.; Gestri, G.; et al. Tissue Macrophages Act as Cellular Chaperones for Vascular Anastomosis Downstream of VEGF-Mediated Endothelial Tip Cell Induction. Blood 2010, 116, 829–840.
- Aurora, A.B.; Porrello, E.R.; Tan, W.; et al. Macrophages Are Required for Neonatal Heart Regeneration. J. Clin. Investig. 2014, 124, 1382–1392.
- Simões, F.C.; Cahill, T.J.; Kenyon, A.; et al. Macrophages Directly Contribute Collagen to Scar Formation during Zebrafish Heart Regeneration and Mouse Heart Repair. Nat. Commun. 2020, 11, 600.
- Chang, M.Y.; Chan, C.K.; Braun, K.R.; et al. Monocyte-to-Macrophage Differentiation: Synthesis and Secretion of a Complex Extracellular Matrix. J. Biol. Chem. 2012, 287, 14122–14135.
- Liu, B.; Zhang, H.-G.; Zhu, Y.; et al. Cardiac Resident Macrophages Are Involved in Hypoxia‑induced Postnatal Cardiomyocyte Proliferation. Mol Med Rep 2017, 15, 3541–3548.
- Ma, F.; Li, Y.; Jia, L.; et al. Macrophage-Stimulated Cardiac Fibroblast Production of IL-6 Is Essential for TGF β/Smad Activation and Cardiac Fibrosis Induced by Angiotensin II. PLoS ONE 2012, 7, e35144.
- Wang, C.; Zhang, C.; Liu, L.; et al. Macrophage-Derived Mir-155-Containing Exosomes Suppress Fibroblast Proliferation and Promote Fibroblast Inflammation during Cardiac Injury. Mol. Ther. 2017, 25, 192–204.
- Yue, Y.; Huang, S.; Wang, L.; et al. M2b Macrophages Regulate Cardiac Fibroblast Activation and Alleviate Cardiac Fibrosis After Reperfusion Injury. Circ. J. 2020, 84, 626–635.
- Wu, L.; Tai, Y.; Hu, S.; et al. Bidirectional Role of Β2-Adrenergic Receptor in Autoimmune Diseases. Front. Pharmacol. 2018, 9, 1313.
- Fu, Q.; Wang, Y.; Yan, C.; et al. Phosphodiesterase in Heart and Vessels: From Physiology to Diseases. Physiol. Rev. 2024, 104, 765–834.
- Hertz, A.L.; Beavo, J.A. Cyclic Nucleotides and Phosphodiesterases in Monocytic Differentiation. In Phosphodiesterases as Drug Targets; Francis, S.H., Conti, M., Houslay, M.D., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 365–390.
- Surdo, N.C.; Berrera, M.; Koschinski, A.; et al. FRET Biosensor Uncovers cAMP Nano-Domains at β-Adrenergic Targets That Dictate Precise Tuning of Cardiac Contractility. Nat. Commun. 2017, 8, 15031.
- Mongillo, M.; Tocchetti, C.G.; Terrin, A.; et al. Compartmentalized Phosphodiesterase-2 Activity Blunts β-Adrenergic Cardiac Inotropy via an NO/cGMP-Dependent Pathway. Circ. Res. 2006, 98, 226–234.
- Xiao, H.; Li, H.; Wang, J.-J.; et al. IL-18 Cleavage Triggers Cardiac Inflammation and Fibrosis upon β-Adrenergic Insult. Eur. Heart J. 2018, 39, 60–69.
- Lyu, J.; Wang, M.; Kang, X.; et al. Macrophage-Mediated Regulation of Catecholamines in Sympathetic Neural Remodeling after Myocardial Infarction. Basic Res. Cardiol. 2020, 115, 56.
- Petkevicius, K.; Bidault, G.; Virtue, S.; et al. Norepinephrine Promotes Triglyceride Storage in Macrophages via Beta2-Adrenergic Receptor Activation. FASEB J. 2021, 35, e21266.
- Liu, W.; Chen, W.; Xie, M.; et al. Traumatic Brain Injury Stimulates Sympathetic Tone-Mediated Bone Marrow Myelopoiesis to Favor Fracture Healing. Signal Transduct. Target. Ther. 2023, 8, 260.
- Ulleryd, M.A.; Bernberg, E.; Yang, L.J.; et al. Metoprolol Reduces Proinflammatory Cytokines and Atherosclerosis in ApoE-/- Mice. BioMed Res. Int. 2014, 2014, e548783.
- Clemente-Moragón, A.; Gomez, M.; Villena-Gutierrez, R.; et al. Metoprolol Exerts a Non-Class Effect against Ischaemia-Reperfusion Injury by Abrogating Exacerbated Inflammation. Eur. Heart J. 2020, 41, 4425–4440.
- Toyoda, S.; Haruyama, A.; Inami, S.; et al. Effects of Carvedilol vs Bisoprolol on Inflammation and Oxidative Stress in Patients with Chronic Heart Failure. J. Cardiol. 2020, 75, 140–147.
- Feuerstein, G.Z.; Ruffolo, R.R. Carvedilol, a Novel Multiple Action Antihypertensive Agent with Antioxidant Activity and the Potential for Myocardial and Vascular Protection. Eur. Heart J. 1995, 16 SupplF, 38–42.
- Zhang, J.; Jiang, P.; Sheng, L.; et al. A Novel Mechanism of Carvedilol Efficacy for Rosacea Treatment: Toll-Like Receptor 2 Inhibition in Macrophages. Front. Immunol. 2021, 12, 609615.
- Cimmino, G.; Ibanez, B.; Giannarelli, C.; et al. Carvedilol Administration in Acute Myocardial Infarction Results in Stronger Inhibition of Early Markers of Left Ventricular Remodeling than Metoprolol. Int. J. Cardiol. 2011, 153, 256–261.
- Udelson, J.E. Ventricular Remodeling in Heart Failure and the Effect of Beta-Blockade. Am. J. Cardiol. 2004, 93, 43–48.
- Apaydin, O.; Altaikyzy, A.; Filosa, A.; et al. Alpha-1 Adrenergic Signaling Drives Cardiac Regeneration via Extracellular Matrix Remodeling Transcriptional Program in Zebrafish Macrophages. Dev. Cell 2023, 58, 2460–2476.
- Bystrom, J.; Evans, I.; Newson, J.; et al. Resolution-Phase Macrophages Possess a Unique Inflammatory Phenotype That Is Controlled by cAMP. Blood 2008, 112, 4117–4127.
- Lima, K.M.; Vago, J.P.; Caux, T.R.; et al. The Resolution of Acute Inflammation Induced by Cyclic AMP Is Dependent on Annexin A1. J. Biol. Chem. 2017, 292, 13758–13773.
- Jiang, H.; Wei, H.; Wang, H.; et al. Zeb1-Induced Metabolic Reprogramming of Glycolysis Is Essential for Macrophage Polarization in Breast Cancer. Cell Death Dis. 2022, 13, 206.
- Ye, J.; Zeng, B.; Zhong, M.; et al. Scutellarin Inhibits Caspase-11 Activation and Pyroptosis in Macrophages via Regulating PKA Signaling. Acta Pharm. Sin. B 2021, 11, 112–126.
- Pan, H.; Lin, Y.; Dou, J.; et al. Wedelolactone Facilitates Ser/Thr Phosphorylation of NLRP3 Dependent on PKA Signalling to Block Inflammasome Activation and Pyroptosis. Cell Prolif 2020, 53, e12868.
- Witwicka, H.; Kobiałka, M.; Siednienko, J.; et al. Expression and Activity of cGMP-Dependent Phosphodiesterases Is up-Regulated by Lipopolysaccharide (LPS) in Rat Peritoneal Macrophages. Biochim. Biophys. Acta 2007, 1773, 209–218.
- Gantner, F.; Kupferschmidt, R.; Schudt, C.; et al. In Vitro Differentiation of Human Monocytes to Macrophages: Change of PDE Profile and Its Relationship to Suppression of Tumour Necrosis Factor-Alpha Release by PDE Inhibitors. Br. J. Pharmacol. 1997, 121, 221–231.
- Down, G.; Siederer, S.; Lim, S.; Daley-Yates, P. Clinical Pharmacology of Cilomilast. Clin. Pharmacokinet. 2006, 45, 217–233.
- Choi, W.S.; Kang, H.S.; Kim, H.J.; et al. Vinpocetine Alleviates Lung Inflammation via Macrophage Inflammatory Protein-1β Inhibition in an Ovalbumin-Induced Allergic Asthma Model. PLoS ONE 2021, 16, e0251012.
- Hsu, C.G.; Fazal, F.; Rahman, A.; et al. Phosphodiesterase 10A Is a Key Mediator of Lung Inflammation. J. Immunol. 2021, 206, 3010–3020.
- Drozdz, M.M.; Doane, A.S.; Alkallas, R.; et al. A Nuclear cAMP Microdomain Suppresses Tumor Growth by Hippo Pathway Inactivation. Cell Rep. 2022, 40, 111412.
- Nash, C.A.; Wei, W.; Irannejad, R.; et al. Golgi Localized Β1-Adrenergic Receptors Stimulate Golgi PI4P Hydrolysis by PLCε to Regulate Cardiac Hypertrophy. Elife 2019, 8, e48167.
- Subramaniam, G.; Schleicher, K.; Kovanich, D.; et al. Integrated Proteomics Unveils Nuclear PDE3A2 as a Regulator of Cardiac Myocyte Hypertrophy. Circ. Res. 2023, 132, 828–848.
- Barbagallo, F.; Xu, B.; Reddy, G.R.; et al. Genetically Encoded Biosensors Reveal PKA Hyperphosphorylation on the Myofilaments in Rabbit Heart Failure. Circ Res 2016, 119, 931–943.
- Benton, K.C.; Wheeler, D.S.; Kurtoglu, B.; et al. Norepinephrine Activates Β1-Adrenergic Receptors at the Inner Nuclear Membrane in Astrocytes. Glia 2022, 70, 1777–1794.
- Bobin, P.; Belacel-Ouari, M.; Bedioune, I.; et al. Cyclic Nucleotide Phosphodiesterases in Heart and Vessels: A Therapeutic Perspective. Arch. Cardiovasc. Dis. 2016, 109, 431–443.


