
Downloads
 Download
Download






This work is licensed under a Creative Commons Attribution 4.0 International License.
Review
Development of Proteasome Inhibitors for Cancer Therapy
Xu Chen † , Xuan Wu † , Linyan Li, and Xiaoming Zhu *
State Key Laboratory of Quality Research in Chinese Medicine, Macau Institute for Applied Research in Medicine and Health, Macau University of Science and Technology, Taipa, Macau SAR, 999078, China
* Correspondence: xmzhu@must.edu.mo
† These authors contributed equally to this work.
Received: 12 January 2024
Accepted: 19 February 2024
Published: 18 March 2024
Abstract: The ubiquitin proteasome system (UPS) is considered a crucial degradation machinery in cellular processes of protein quality control and homeostasis. Dysregulation of the UPS is closely associated with many diseases. The proteasome is a key core component of the UPS, which can prevent the accumulation of misfolded proteins and regulate various cellular processes such as cell cycle, apoptosis, and immune responses. In the past two decades, a total of three proteasome inhibitors have been approved for the treatment of hematological malignancies, including bortezomib, carfilzomib, and ixazomib. Additionally, accumulating reports have suggested that some natural product-derived proteasome inhibitors have been developed as anti-cancer drug candidates. In this review, we summarize the development of proteasome inhibitors as well as the mechanisms involved, clinical application progress, and drug resistance. The natural products of proteasome inhibitors and their future perspectives will also be discussed.
Keywords:
Ubiquitin proteasome system proteasome inhibitors natural products1. Introduction
Proteostasis is a key requirement for cell metabolism, organelle biogenesis, and stress adaptation [1]. A major challenge in proteostasis is to prevent the deleterious consequences of unfolded, misfolded, or damaged proteins that severely disrupt cellular function and are associated with aging and age-related diseases, such as neurodegenerative disorders, cancers, and immunologic and metabolic diseases [2,3]. Two major quality control systems responsible for the degradation of proteins in all eukaryotic cells are the ubiquitin proteasome system (UPS) and the autophagy-lysosome pathway (ALP). These systems are highly regulated and are responsible for maintaining protein homeostasis and adapting to environmental changes through the degradation of multiple proteins [4,5]. UPS is the main pathway for protein degradation [6], primarily degrading single unfolded polypeptides that have access to the narrow channel of the proteasome, which is a major proteolytic pathway for short-lived proteins, misfolded proteins, and damaged proteins [7].
Inhibition of the proteasome has become a vital target for drug development in cancer and other diseases in recent years [8]. Proteasome inhibitors are currently one of the most important chemotherapeutic drugs for the treatment of multiple myeloma (MM) and mantle cell lymphoma (MCL) [9]. In 2003, the US Food and Drug Administration (FDA) approved the first proteasome inhibitor, bortezomib, followed by carfilzomib and ixazomib in 2012 and 2015, respectively [10]. Additionally, several natural compounds and their derivatives have recently been identified as proteasome inhibitors. Owing to their unique chemical diversity, natural products possess a diversity of biological activities and pharmaceutical properties. In the search for new pharmacologically active substances, natural products from various sources have been shown to play an important role in the treatment of many diseases. Reports indicate that polyphenols like epigallocatechin gallate (EGCG) [11], quercetin [12], terpenoids such as celastrol [13] and petrosapongiolide M [14] have been proven to be proteasome inhibitors.
In this review, we describe and discuss: 1) the UPS process and related molecular mechanisms; 2) the clinically used as well as other potential proteasome inhibitors; 3) The molecular mechanism by which they inhibit the proteasome; 4) natural products for proteasome inhibition and their future perspectives.
2. General Overview of Ubiquitin Proteasome System
2.1. UPS
The proteasome system regulates cellular functions and removes damaged, misfolded, or redundant proteins from cells in a well-orchestrated manner [15]. This pathway consists of two highly coordinated steps, including ubiquitination of the substrate proteins and their degradation by the proteasome ( Figure 1). To guarantee that the proteasome accurately recognizes the protein to be degraded, ubiquitin must first be attached to the target protein. Ubiquitin is a 76 amino acid protein with various functions, especially protein degradation [15]. Polyubiquitination is mediated by a cascade reaction including a series of enzymes (E1, E2, and E3) that sequentially transmit ubiquitin molecules to the corresponding cellular targets [16]. The ubiquitinated protein is recognized by a special regulatory subunit on the proteasome complex to mediate deubiquitination, and the ubiquitin chain is removed for subsequent recycling. The protein is then unfolded by the proteasome complex and transferred to the internal cleavage into peptide products [15].
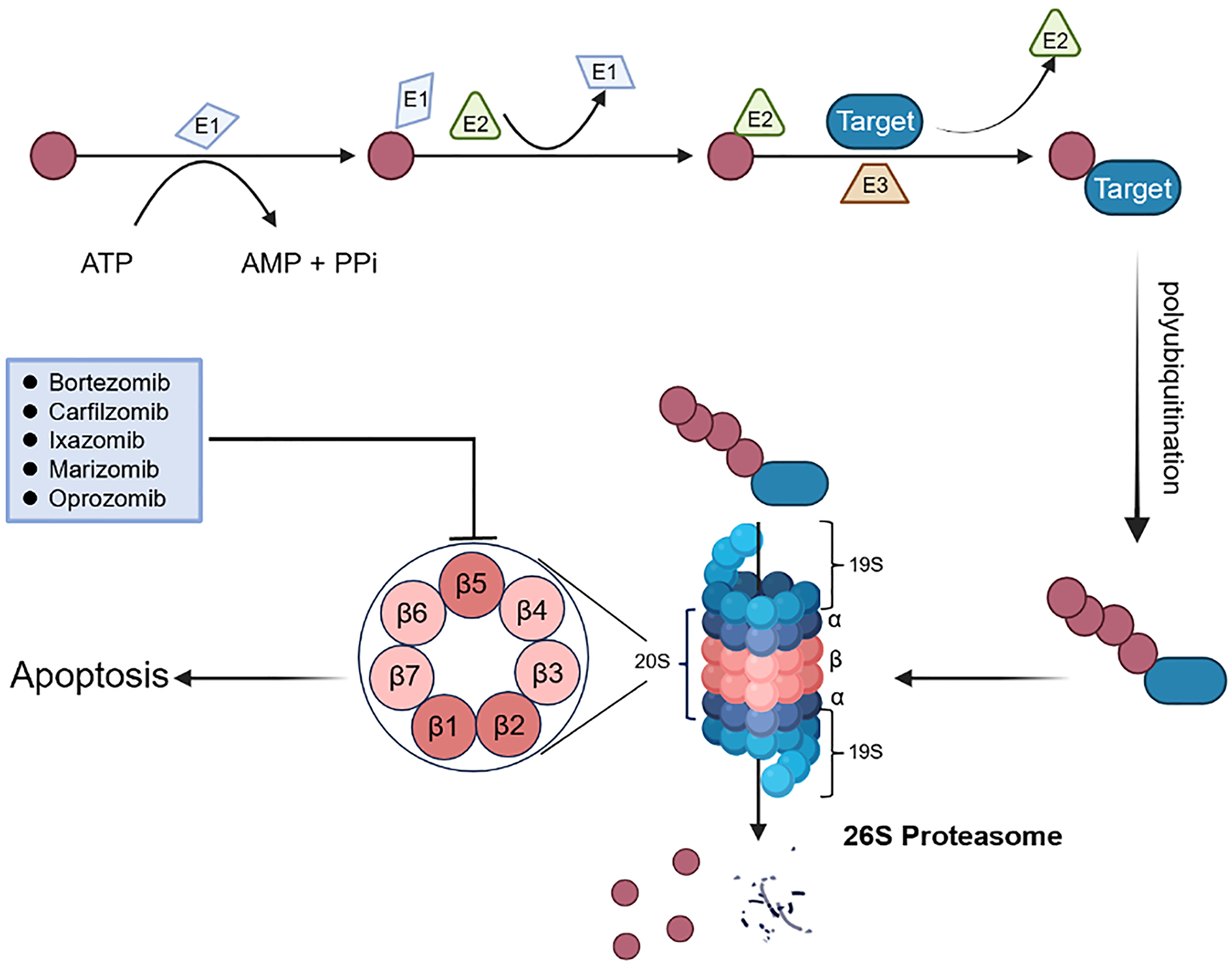
Figure 1. The overview of UPS and proteasome inhibitors. Ubiquitin (Ub) is activated by the activating enzyme E1, Ub is then transferred to the conjugating enzyme E2. The ligase E3 enzyme attaches Ub to the target protein (substrate) and a substrate with at least four Ub moieties is recognized by the proteasome for degradation.
2.2. Composition of the Proteasome
The proteasome complex is not static but varies in many ways due to compositional changes in structural subunits, catalytic subunits, post-translational modifications, and regulatory subunits [17]. Generally, different forms of proteasome could be regulated according to the conditions in the cells, and the proteasome forms can also differ among cell types and tissues with different functions [17].
The proteasome mainly refers to the 26S proteasome, which catalyzes the degradation of the vast majority (at least 80%) of proteins in growing mammalian cells, operating in a ubiquitin- and ATP-dependent manner [9]. The 26S proteasome is a multiprotein complex consisting of a catalytic core 20S in the middle catalytic position and one or two 19S regulatory subunits at either end ( Figure 1) [18]. The 20S proteasome is the most important component of the ubiquitin-dependent protein degradation pathway [19]. In some cases, the 20S core can also act alone, leading to the degradation of ubiquitin-independent proteins [16]. The 20S core particle is a barrel-shaped structure composed of four heptameric rings formed by 14 α and 14 β subunits, and the two outer and inner rings are seven α subunits (α1-7) and β subunits (β1-7), respectively. The outer α subunits are configured to form a barrier by its N-terminal to prevent proteins from entering and leaving the core. The protease activity of the proteasome is attributed to three catalytic sites with distinct cleavage specificities, including caspase-like (β1), trypsin-like (β2), and chymotrypsin-like (β5) activities. Among them, the chymotrypsin-like site on β5 is the primary target of proteasome inhibitors as it is the main rate-limiting factor in the process of protein degradation [20]. In addition, the immunoproteasome versions of the catalytic sites are β1i, β2i, and β5i subunits. “i” stands for immunoproteasome, the predominant form of proteasome in corresponding antigen-presenting cells of the body’s immune system [21]. The remaining four inactive β-protease subunits are homologues of the three active subunits mentioned above [21]. In most occasions, 20S core particles coexist with three kinds of regulatory complexes, 19S, 11S and PA200 [22].
2.3. Mechanism of Proteasome Inhibitors-Mediated Cytotoxicity
The UPS is involved in a wide range of processes, participating in various mechanisms of cell death, and affecting many pathways in tumor cells. Tumor cells undergo abnormal changes in intracellular metabolism due to unlimited proliferation, leading to a higher occurrence of misfolded proteins during the process of massive protein synthesis compared to normal cells [23]. Under excessively high metabolic levels, the quantity of proteins that require degradation within cells also rises significantly. Therefore, proteasome activity is maintained at a high level in numerous tumor cells. Inhibition of proteasome activity with proteasome inhibitors often leads to the inability of tumor cells to metabolize intracellular proteins in time, causing an imbalance in intracellular metabolism, and ultimately leading to the demise of tumor cells [24]. Proteasome inhibitors have been shown to induce apoptosis through multiple pathways. The main mechanisms by which proteasome inhibition induces apoptosis and cell death are discussed below.
2.3.1. Endoplasmic Reticulum (ER) Stress and Oxidative Stress
Malignant tumors are characterized by alterations in the mTOR signaling and growth signaling pathways, which generally reflect significant increases in the rate of protein synthesis. Excessive protein synthesis in tumor cells overloads the ER’s ability to fold proteins, leading to acute proteotoxic stress and ultimate ER stress. Under the ER stress, misfolded proteins in cells are primarily degraded by the UPS to maintain their stability. Tumor cells are more susceptible to proteasome inhibitors than healthy cells because of the higher rates of translation-induced protein misfolding in tumor cells. Numerous studies have demonstrated that in vitro administering different proteasome inhibitors to tumor cells can produce acute ER stress and lead to apoptosis [25‒28]. Notably, oxidative stress makes proteins vulnerable to oxidative changes when they are folded in the ER. Although the connection between oxidative stress and proteasome inhibitors is unclear, some studies have demonstrated that inhibition of ROS generation by antioxidants can significantly reduce proteasome inhibitor-induced apoptosis. This suggests that oxidative stress plays a key role in triggering apoptosis through UPS inhibition [29,30].
2.3.2. NF-κB Signal Pathway
NF-κB is an anti-apoptotic factor closely related to tumor growth and an anti-tumor pathway associated with proteasome inhibitors [31]. The IκB kinase inactivates NF-κB by inhibiting the p65-p50 heterodimer. After stimulating cells with antigens, cytokines, viruses, etc., IκB is modified by phosphorylation and ubiquitination, and then degraded by the proteasome to release NF-κB. The released NF-κB is subsequently translocated into the nucleus. NF-κB binds to genes encoding anti-apoptotic factors and promotes the transcription and expression of anti-apoptotic proteins. Proteasome inhibitors can prevent the degradation of IκB by inhibiting the activity of the UPS, thereby inhibiting the activation of the anti-apoptotic factor NF-κB and exerting an anti-tumor effect [32].
2.3.3. Cell Cycle Regulation
Through the detection, interaction, and ubiquitination or deubiquitination of important proteins, the UPS guarantees a unidirectional way throughout the cell cycle and coordinates movement between each phase of the cell cycle. Therefore, the UPS is crucial for controlling cell cycle progression. The activity of cyclin-dependent kinases (CDKs) is controlled by ubiquitin-mediated proteolysis of important regulators such as cyclins, the CDK inhibitors, or other kinases and phosphatases, and is responsible for cell cycle changes. Since most cell cycle regulators are the UPS substrates, proteasome inhibition results in uncontrolled cell cycle progression and growth arrest in the G1 and G2 stages of the cell cycle [33]. The p53 gene is a human tumor suppressor gene, which encodes the p53 protein with a strong tumor suppressor effect and has also attracted considerable research and interest for its role in cell cycle control. The ability of p53 protein to arrest the cell cycle is due to its indirect downregulation of the numerous gene expressions required for cell division cycle progression. The p53 protein has the physiological function of activating DNA repair proteins and preventing cell growth. Sequence variants or deletion of p53 gene are found in most cancer patients. Under normal circumstances, the half-life of the p53 protein is very short, and blocking proteasome activity is beneficial to reduce the degradation of p53 protein and increase its level rapidly [34].
3. Small Molecule Proteasome Inhibitors for Cancer Therapy
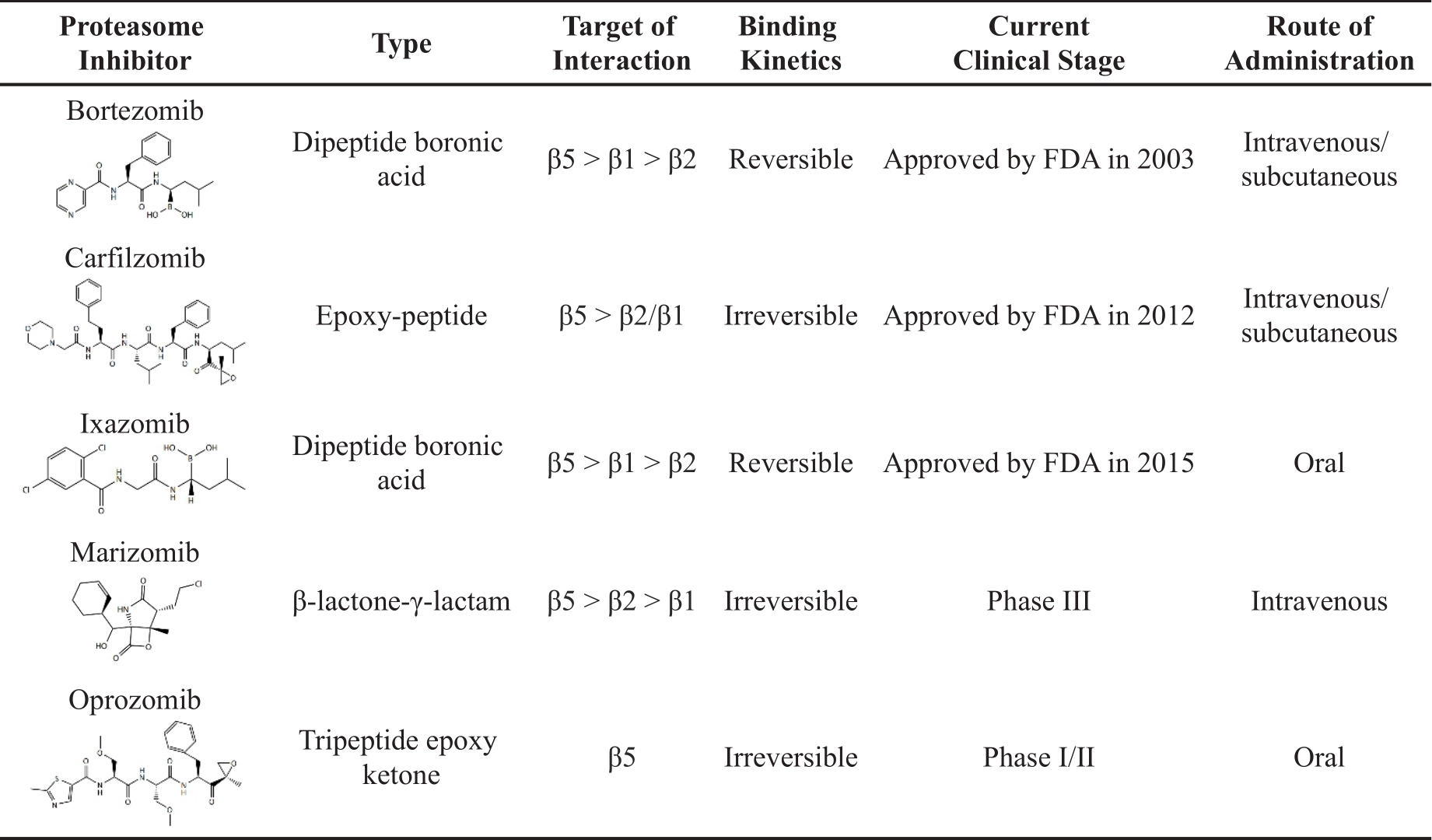
Owing to the role of the UPS in protein degradation, proteasome inhibitors were initially developed as agents with potential benefit in preventing cancer-related cachexia. Numerous preclinical studies have subsequently shown that small-molecule proteasome inhibitors have the potential to be used as chemotherapy drugs because they could induce apoptosis in cultured cell lines and mouse cancer models. Research on proteasome inhibitors is mainly aimed at the inhibition of the proteasome 20S proteolytic core, and the proteasome inhibitors function by forming reversible or irreversible covalent compounds with the threonine active site of the proteasome [35]. According to their different chemical structures, proteasome inhibitors can be categorized into peptides and non-peptides. Peptides include peptide boronates, peptide epoxyketones, peptide aldehydes, peptide heterocycles, peptide vinyl sulfones and cyclopeptides, while non-peptides include β-lactones and others. To date, the proteasome inhibitors approved by the FDA are bortezomib, carfilzomib, and ixazomib for the treatment of MM or MCL [36]. Other novel proteasome inhibitors, such as marizomib and oprozomib, are currently under preclinical trials ( Table 1).
Table 1. Proteasome inhibitors approved or in clinical trials.
3.1. Bortezomib
Bortezomib is the first proteasome inhibitor, approved by the FDA in 2003 [37]. The structure of bortezomib consists of a peptide-like backbone and a boronic easter part. This boronic ester part binds tightly to the threonine catalytic site in the 20S proteasome, resulting in a slow and reversible inhibition of its activity. Specifically, bortezomib mainly inhibits the β5 activity of the proteasome, while the inhibition of β1 and β2 activities is weaker. By inhibiting the proteasome, bortezomib disrupts the degradation of proteins within cells and can induce cell cycle arrest and cell death in cancer cells. It is typically utilized in the first-line treatment of patients with MM or MCL [19].
It is reported that bortezomib can effectively inhibit the 20S proteasome by targeting threonine residues (Ki = 0.6 nM) [23]. The IC50 value of bortezomib against U266 MM cells is 30 nM [38]. Bortezomib can also induce apoptosis in MCL cell lines [39]. In addition, studies have reported the effect of bortezomib on different lymphoma cell lines, with EC50 values ranging from 6 nM (DHL-7 cells) to 25 nM (DHL-4 cells) [40]. It is worth noting that bortezomib has been shown to induce apoptosis and inhibit the growth of human myeloma cells. Bortezomib possesses the IC50 value of 30 nM in human MM RPMI 8226 MM cells [41]. In subsequent animal experiments, tumor growth was significantly inhibited and even completely regressed in bortezomib-treated myeloma mice. In addition, bortezomib has also demonstrated potent anti-tumor efficacy in many preclinical solid tumor mouse models, such as lung, head and neck, breast, and melanoma xenograft mouse models [23,42].
Of note, clinical trials have utilized bortezomib alongside other anti-cancer medications to address MM and MCL [43]. Most patients receive a clinical regimen consisting of immunomodulators and proteasome inhibitors, typically lenalidomide and bortezomib combined with dexamethasone. Phase I-III trials have demonstrated that this combination is associated with favorable response rates, manageable toxicity, and clinical benefit. The Southwest Oncology Group S0777 phase III research shows that adding bortezomib to lenalidomide and dexamethasone dramatically improves the prognosis of patients with previously untreated MM. Survival rates were improved with an increase of 11 months in overall survival and 13 months in progression-free survival (PFS) [44]. In addition, the VcR-CVAD regimen (a combination of bortezomib, rituximab, cyclophosphamide, vincristine, doxorubicin, and dexamethasone) is effective and well tolerated in patients with primary MCL. The complete response rate, overall response rate (ORR), and 3-year PFS were 68%, 95%, and 72%, respectively [45,46]. Clinically representative treatment options include VD (bortezomib plus dexamethasone) [47] and VR-CAP (bortezomib, rituximab, cyclophosphamide, doxorubicin, and prednisone) [48].
3.2. Carfilzomib
Bortezomib has a limited range of applications due to its severe toxicity and drug resistance. The natural substance epoxomicin underwent structural alteration to create the next generation of proteasome inhibitor carfilzomib. The FDA subsequently authorized carfilzomib in 2012. Carfilzomib is an irreversible proteasome inhibitor, and its epoxy ketone can interact with the threonine active site of the proteasome to establish a covalent link, which makes it different from bortezomib. Epoxyketones can interact with the N-terminal threonine of the proteasome active site, which is similar to the mode of action of borates (such as bortezomib and ixazomib). However, unlike reversibly bound borates, the irreversible covalent bonding of epoxyketones with an N-terminal threonine can inhibit proteasome longer. Studies have demonstrated that carfilzomib is superior to bortezomib in preclinical and clinical contexts [49,50]. Carfilzomib outperforms bortezomib in enhancing Janus kinase phosphorylation and/or caspase activity in acute lymphoblastic leukemia and MM cell lines [50]. Carfilzomib also shows a promising effect on inducing apoptosis in bortezomib-resistant MM cells [50].
Carfilzomib exhibits a preferential inhibitory effect on β5 activity, effectively reducing it by more than 80% at concentrations of 10 nM [50]. The carfilzomib effectively inhibits the viability of RPMI 8226 cells with the IC50 value of 5 nM. Additionally, carfilzomib can overcome dexamethasone resistance in MM1.R cells with the IC50 of 15.2 nM, which is lower than the value of 29.3 nM in parental MM1.S cells [50].
During the clinical phase I trial, carfilzomib was given intravenously for patients with MM, Hodgkin’s disease and non-Hodgkin’s lymphoma every 14 days on day 1‒5 and the dose ranged from 1.2 to 20 mg/m2 over 1‒2 min. The ORR for MM patients was 7.1% and the clinical benefit response rate (CBR) was 14.3% [51]. The dose of carfilzomib was increased to 20 mg/m2 in the first cycle and 27 mg/m2 in the succeeding cycles of Phase II studies PX-171-003 and PX-171-004, in which patients with MM received extensive pretreatment with bortezomib. Higher ORR and CBR were seen in both trials compared to the PX-171-001 trial, with ORRs of 17.1% and 23.7%, and CBRs of 31.4% and 37%, respectively [52,53]. Furthermore, the findings of its phase III clinical study demonstrated that carfilzomib is effective in patients who have had a relapse and are resistant to bortezomib [54,55]. Clinical evidence from phase I and II trials evaluating individuals with advanced MM suggests that combining carfilzomib and dexamethasone may be a beneficial therapy option for those with relapsed/refractory MM (RRMM) [56,57]. Although carfilzomib has been demonstrated to be superior to first-generation proteasome inhibitor bortezomib, there are still limitations in terms of its poor efficacy in patients with solid tumors [58].
3.3. Ixazomib
Ixazomib is the first oral reversible proteasome inhibitor, approved by the FDA in 2015. Its active form consists of a borate moiety [59]. Ixazomib citrate is a prodrug that is rapidly hydrolyzed to bioactive ixazomib under physiological conditions. Carfilzomib or ixazomib often shows excellent efficacy when patients are resistant to bortezomib. Compared with bortezomib, ixazomib has better pharmacokinetic and pharmacodynamic properties and exhibits more potent anti-tumor activity in hematologic xenograft and solid tumor mouse models [60].
Preclinical studies have shown that ixazomib selectively inhibits β5 activity of 20S proteasome with an IC50 value of 3.4 nM and suppresses β1 activity at a higher concentration (IC50 value of 31 nM) [60]. This experiment also conducted cell viability studies in a variety of mammalian cell lines to detect the anti-proliferative effect of ixazomib in vitro. In addition, ixazomib citrate prevents all three proteolytic sites (β5, β2, β1) in MM cells [61]. It is reported that ixazomib efficiently inhibits the growth of MG-63 and Saos-2 cells in a time- and dose-dependent manner with IC50 values of 0.4 μM and 0.8 μM, respectively [61]. Ixazomib (11 mg/kg) effectively reduces the proliferation of the MM cells in vivo and increases the survival of the human plasmacytoma MM.1S xenograft mice model [62]. Clinically, ixazomib combined with dexamethasone and lenalidomide can increase PFS in patients with RRMM [63]. In addition, in vitro studies have shown that ixazomib can induce apoptosis in both bortezomib- and lenalidomide-resistant cell lines [62]. Notably, the main side effects of ixazomib are skin rash and gastrointestinal adverse events [60].
3.4. Marizomib
Salinosporamide A, also known as marizomib, is a natural substance isolated from the bacteria Salinispora in marine sediments and acts as an irreversible proteasome inhibitor [10]. Studies have shown that marizomib can inhibit the activities of β5, β1, and β2 of the 20S proteasome, with IC50 values of 3.5, 28, and 430 nM, respectively [64]. Furthermore, marizomib significantly inhibits the proteasome activity on U-251 and D-54 cells [65]. The immunostaining analysis of MM tumors treated with the combination of marizomib and bortezomib demonstrates significant growth inhibition, induction of apoptosis, and concurrent angiogenesis [38]. Marizomib also significantly slows tumor development in a human MM xenograft murine model when administered intravenously twice a week at a dose of 0.15 mg/kg for three weeks [66].
3.5. Oprozomib
Oprozomib, a tripeptide analog of carfilzomib, is an orally bioavailable proteasome inhibitor in clinical trials for the treatment of newly diagnosed and RRMM with severe gastrointestinal reactions [67]. Oprozomib shows anti-cancer efficacy equivalent to carfilzomib in preclinical studies [68]. The IC50 value of oprozomib is 36 nM [59]. It is worth noting that the oral oprozomib-pomalidomide-dexamethasone combination has also shown promise in clinical studies for treating individuals with RRMM [69].
Currently, several other compounds are being studied in clinical trials. Zetomipzomib and M-3258 are immunoproteasome inhibitors that have entered clinical trials with higher solubility [70,71]. ACU-D1 is a novel proteasome inhibitor for the treatment of moderate to severe rosacea, which can reduce inflammatory lesions and erythema in affected patients. These ongoing clinical trials and investigations hold promise for expanding the use of proteasome inhibitors beyond their current indications, offering new treatment options and hope for patients across various disease areas.
4. Natural Products as Proteasome Inhibitors
The use of natural products in preventing and treating diseases has a long history. Recent research indicates that various natural products play a role in engaging the UPS through their pharmacological actions [72]. Several natural compounds and their derivatives have been identified as proteasome inhibitors in recent years ( Figure 2). The discovery and identification of these molecules have the potential to pave the way for the development of powerful anti-cancer drugs. These medications can effectively combat the negative effects and resistance mechanisms that are commonly observed with currently approved proteasome inhibitors. This breakthrough could significantly enhance clinical advancements in the field and offer more effective treatment options for cancer patients.
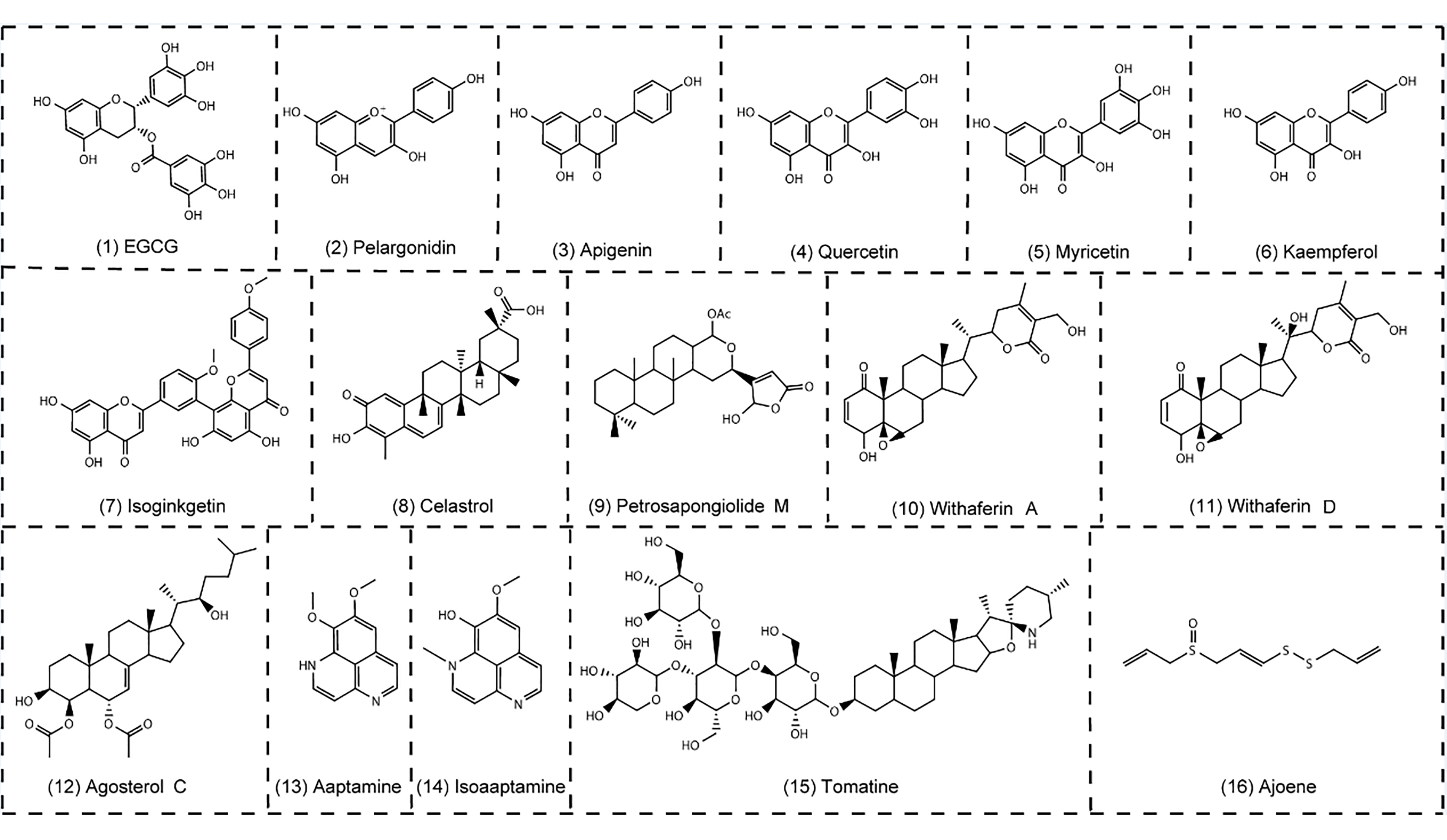
Figure 2. Natural products as proteasome inhibitors. (1)‒(7), polyphenols; (8)‒(12), terpenoids and steroid; (13)‒(15), alkaloids; (16) ajoene.
4.1. Polyphenols
Natural polyphenols are widely found in cocoa beans, tea, vegetables, fruits, as well as some Chinese medicinal herbs. They are a class of plant compounds containing a variety of phenolic structural units with potent anti-oxidative and anti-inflammatory properties. Polyphenols are composed of flavonoids (such as flavanols, anthocyanidins, anthocyanins, isoflavones, etc.) and non-flavonoids (such as phenolic acids, stilbenes, and lignans). The anti-cancer effects of naturally occurring polyphenols have become a hot topic in many studies over the past two decades [73].
The EGCG, an active ingredient of green tea, is one of the prominent phenolic proteasome inhibitors [73]. The EGCG has an inhibitory effect on the β5 activity of the 20S proteasome [11,74‒75]. The IC50 value of the EGCG is 86 nM [76]. Studies have shown that anthocyanins and their aglycons have inhibitory effects on the proteasome [12]. Apigenin, a flavonoid found in a variety of fruits and vegetables, has been shown to suppress the activity of the proteasome and cause apoptosis in leukemia Jurkat T cells [12,77‒78]. The IC50 value of apigenin is 1.8 μM [12] In addition to apigenin, other dietary flavonoids such as quercetin, myricetin, and kaempferol can effectively inhibit the β5 activity [12,80]. Studies have shown that kaempferol is six times less potent than apigenin, with the IC50 value of 10.5 μM. It is speculated that hydroxyl groups at specific positions contribute to the ability of these flavonoids to inhibit the proteasome [12]. However, although having the same C3 hydroxyl group, quercetin exhibits superior proteasome inhibitory activity to myricetin, with the IC50 values of 3.5 μM and 10 μM, respectively [12]. Isoginkgetin is a natural biflavonoid proteasome inhibitor that reduces the 20S proteasome by greater than 50% at a dose of 30 μM [80].
4.2. Terpenoids and steroid
A class of natural chemicals with a four-ring structure often found in plants are called terpenoids and steroids. Triterpenoids are a powerful group of phytochemicals derived from plant foods and herbs, which have been shown in several reports to exhibit chemotherapeutic effects in cell lines and animal models [81‒83].
Celastrol is an active compound isolated from the root of the “Thunder of God Vine” ( Tripterygium wilfordii) used in traditional Chinese medicine. Some results indicate that celastrol is a natural proteasome inhibitor with great potential for cancer prevention and treatment. Celastrol can inhibit β5 activity of purified 20S proteasome with the IC50 value of 2.5 μM in vitro [13,84‒87]. Petrosapongiolide M is a natural terpene with anti-inflammatory properties. A pharmacological investigation reveals that Petrosapongiolide M has a significant inhibition of the β1 and β5 activities with the IC50 values of 0.85 ± 0.15 μM and 0.64 ± 0.15 μM, respectively [14].
Oxygenated steroids represent a class of proteasome inhibitors, among which withaferins are the most prominent anti-tumor compounds found in a steroidal lactone purified from the medicinal plant “Indian Winter Cherry” ( Withania somnifera) [88‒90]. Withanolides are a group of naturally occurring C28-steroidal lactones. Steroids have a particular configuration of four linked cycloalkane ring structures, three cyclohexane rings, and one cyclopentane ring. Withaferin A potently inhibits the β5 activity of the purified rabbit 20S proteasome with the IC50 value of 4.5 μM, which highlights the potential use of this natural product for cancer treatment or prevention [88‒90]. The agosterols are a large group of natural products consisting of more than a dozen high-oxygen steroids [91]. Agosterol C has the IC50 value of 10 μg/mL for proteasome inhibition, making the substance more desirable in preventing tumor cells from developing multidrug resistance [92].
4.3. Alkaloids
Studies have shown that aaptamine, isoaaptamine, and demethylaaptamine isolated from the marine sponge Aaptos suberitoides collected in Indonesia can act as proteasome inhibitors. These substances inhibit the β5 and β1 activities of the proteasome with IC50 values of 1.6‒4.6 μg/mL, but the inhibitory effect on β2 activity is not obvious [93]. Steroidal alkaloids are a class of natural products that occur in several species of the Solanaceae family. In the case of the tomato plant ( Lycopersicon esculentum Mill.), tomatine and its aglycone, tomatidine, are the most representative molecules. Tomatine has been reported to promote the upregulation of nuclear apoptosis inducing factors in neuroblastoma cells, as well as inhibit 20S proteasome activity with the IC50 value of 2 μM [94].
4.4. Others
In addition to the ones summarized above, numerous other natural products have inhibitory effects on the proteasome. Ajoene has been reported to affect proteasome function and activity both in vitro and in vivo, its role in the prevention and treatment of cancer is receiving increasing attention. Furthermore, epoxyphomalins A and B, a new family of natural products that are from a marine-derived fungus, are also a class of proteasome inhibitors. They are found to be cytotoxic, particularly against prostate PC3M cells and bladder BXF 1218 L cells with the IC50 value of 0.72 µM and 1.43 µM, respectively [95].
5. Conclusion and Perspective
The UPS plays an important role in the onset and progression of cancer and in the development of chemotherapy resistance. Clinically proteasome inhibitors are mainly used in the treatment of MM and MCL with ideal therapeutic effects [96]. It should be noted that the success of proteasome inhibitors in treating hematologic malignancies has not yet been reached in the therapy of solid tumors [58]. Despite of the encouraging results about suppressing the growth of solid tumors in animal studies, the proteasome inhibitors show limited efficacy in several reported clinical trials [97]. This may be related to the low estimated abundance of immunoproteasome in solid tumors [98]. Moreover, the clinical applications of proteasome inhibitors still have some challenges including drug resistance and severe side effects.
To address these issues, there is an urgent need to develop novel proteasome inhibitors with different targets. Small molecule proteasome inhibitors are still in development. Natural products as the source of drug discovery also show great potential as the next generation of proteasome inhibitors. On the other side, inspired by the function of UPS, targeted protein degradation induced by proteolysis-targeting chimeras (PROTACs) have attracted more and more attentions, as it may provide new possibilities for drug discovery and provide tangible therapeutic benefits [99].
Author Contributions: writing-original draft preparation, Xu Chen, Xuan Wu; writing-review and editing, Xu Chen, Xuan Wu, Linyan Li; supervision and funding acquisition, Xiaoming Zhu.
Funding: This study was funded by The Science and Technology Development Fund, Macau SAR (File No. 0035/2022/A1, 006/2023/SKL).
Institutional Review Board Statement: Not applicable.
Informed Consent Statement: Not applicable.
Data Availability Statement: Not applicable.
Conflicts of Interest: The authors declare no conflict of interest.

References
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435‒464. doi: 10.1146/annurev-biochem-060614-033955
- Hipp, M.S.;P. Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421‒435. doi: 10.1038/s41580-019-0101-y
- Liang, R.; Tan, H.B.; Jin, H.J.; et al. The tumour-promoting role of protein homeostasis: Implications for cancer immunotherapy. Cancer Lett. 2023, 573, 216354. doi: 10.1016/j.canlet.2023.216354
- Dikic, I. Proteasomal and autophagic degradation systems. Annu. Rev. Biochem. 2017, 86, 193‒224. doi: 10.1146/annurev-biochem-061516-044908
- Chen, J.L.; Wu, X.; Yin, D.; et al. Autophagy inhibitors for cancer therapy: small molecules and nanomedicines. Pharmacol. Ther. 2023, 249, 108485. doi: 10.1016/j.pharmthera.2023.108485
- Kwon, Y.T.; Ciechanover, A. The ubiquitin code in the ubiquitin-proteasome system and autophagy. Trends Biochem. Sci. 2017, 42, 873‒886. doi: 10.1016/j.tibs.2017.09.002
- Finley, D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 2009, 78, 477‒513. doi: 10.1146/annurev.biochem.78.081507.101607
- Huang, H.; Weng, H.; Dong, B.; et al. Oridonin triggers chaperon-mediated proteasomal degradation of BCR-ABL in leukemia. Sci. Rep. 2017, 27, 415‒425. doi: 10.1038/srep41525
- Narayanan, S.; Cai, C.Y.; Assaraf, Y.G.; et al. Targeting the ubiquitin-proteasome pathway to overcome anti-cancer drug resistance. Drug Resistance Updates. 2020, 48, 100663. doi: 10.1016/j.drup.2019.100663
- Fricker, L.D. Proteasome inhibitor drugs. Annu. Rev. Pharmacol. Toxicol., 2020, 60, 457‒476. doi: 10.1146/annurev-pharmtox-010919-023603
- Dou, Q.P.; Landis-Piwowar, K.R.; Chen, D.; et al. Green tea polyphenols as a natural tumour cell proteasome inhibitor. Inflammopharmacology 2008, 16, 208‒212. doi: 10.1007/s10787-008-8017-8
- Chen, D.; Daniel, K.G.; Chen, M.S.; et al. Dietary flavonoids as proteasome inhibitors and apoptosis inducers in human leukemia cells. Biochem. Pharmacol. 2005, 69, 1421‒1432. doi: 10.1016/j.bcp.2005.02.022
- Yang, H.; Chen, D.; Cui, Q.C.; et al. Celastrol, a triterpene extracted from the Chinese “Thunder of god vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006, 66, 4758‒4765. doi: 10.1158/0008-5472.CAN-05-4529
- Margarucci, L.; Monti, M.C.; Tosco, A.; et al. Chemical proteomics discloses petrosapongiolide M, an antiinflammatory marine sesterterpene, as a proteasome inhibitor. Angew. Chem., Int. Ed. Engl. 2010, 49, 3960‒3963. doi: 10.1002/anie.200907153
- Collins, G.A.; Goldberg, A.L. The logic of the 26S proteasome. Cell 2017, 169, 792‒806. doi: 10.1016/j.cell.2017.04.023
- Nunes, A.T.; Annunziata, C.M. Proteasome inhibitors: structure and function. Semin. Oncol. 2017, 44, 377‒380. doi: 10.1053/j.seminoncol.2018.01.004
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; et al. Structure and function of the 26S proteasome. Annu. Rev. Biochem. 2018, 87, 697‒724. doi: 10.1146/annurev-biochem-062917-011931
- Adams, J. The proteasome: structure, function, and role in the cell. Cancer Treat. Rev. 2003, 29, 3‒9. doi: 10.1016/S0305-7372(03)00081-1
- Nussbaum, A.K.; Dick, T.P.; Keilholz, W.; et al. Cleavage motifs of the yeast 20S proteasome beta subunits deduced from digests of enolase 1. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 12504‒12509. doi: 10.1073/pnas.95.21.12504
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417‒433. doi: 10.1038/nrclinonc.2016.206
- Murata, S.; Takahama, Y.; Kasahara, M.; et al. The immunoproteasome and thymoproteasome: functions, evolution and human disease. Nat. Immunol. 2018, 19, 923‒931. doi: 10.1038/s41590-018-0186-z
- Deshmukh, F.K.; Yaffe, D.; Olshina, M.A.; et al. The contribution of the 20S proteasome to proteostasis. Biomolecules 2019, 9, 190. doi: 10.3390/biom9050190
- Adams, J.; Palombella, V.J.; Sausville, E.A.; et al. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res. 1999, 59, 2615‒2622.
- Yamamoto, Y.; Gaynor, R.B. Therapeutic potential of inhibition of the NF-kappaB pathway in the treatment of inflammation and cancer. J. Clin. Invest. 2001, 107, 135‒142. doi: 10.1172/JCI11914
- Adams, J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell 2004, 5, 417‒421. doi: 10.1016/S1535-6108(04)00120-5
- Hartl, F.U.; Hayer-Hartl, M. Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 2009, 16, 574‒581. doi: 10.1038/nsmb.1591
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 2021, 21, 71‒88. doi: 10.1038/s41568-020-00312-2
- Nikesitch, N.; Lee, J.M.; Ling, S.; et al. Endoplasmic reticulum stress in the development of multiple myeloma and drug resistance. Clin. Transl. Immunol. 2018, 7, e1007. doi: 10.1002/cti2.1007
- Brnjic, S.; Mazurkiewicz, M.; Fryknäs, M.; et al. Induction of tumor cell apoptosis by a proteasome deubiquitinase inhibitor is associated with oxidative stress. Antioxid. Redox Signaling 2014, 21, 2271‒2285. doi: 10.1089/ars.2013.5322
- Fribley, A.; Wang, C.Y. Proteasome inhibitor induces apoptosis through induction of endoplasmic reticulum stress. Cancer Biol. Ther. 2006, 5, 745‒748. doi: 10.4161/cbt.5.7.2971
- Lee, K.H.; Lee, J.; Woo, J.; et al. Proteasome inhibitor-induced IkappaB/NF-kappaB activation is mediated by Nrf2-dependent light chain 3B induction in lung cancer cells. Mol. Cells 2018,41, 1008‒1015.
- Kim, C.; Lee, J.H.; Ko, J.H.; et al. Formononetin regulates multiple oncogenic signaling cascades and enhances sensitivity to bortezomib in a multiple myeloma mouse model. Biomolecules 2019, 9, 262. doi: 10.3390/biom9070262
- Zou, T.; Lin, Z. The involvement of ubiquitination machinery in cell cycle regulation and cancer progression. Int. J. Mol. Sci. 2021, 22, 5754. doi: 10.3390/ijms22115754
- Halasi, M.; Pandit, B.; Gartel, A.L. Proteasome inhibitors suppress the protein expression of mutant p53. Cell Cycle 2014, 13, 3202‒3206. doi: 10.4161/15384101.2014.950132
- Thibaudeau, T.A.; Smith, D.M. A practical review of proteasome pharmacology. Pharmacol. Rev. 2019, 71, 170‒197. doi: 10.1124/pr.117.015370
- Wu, P.; Oren, O.; Gertz, M.A.; et al. Proteasome inhibitor-related cardiotoxicity: mechanisms, diagnosis, and management. Curr. Oncol. Rep. 2020, 22, 66. doi: 10.1007/s11912-020-00931-w
- Orlowski, R.Z.; Stinchcombe, T.E.; Mitchell, B.S.; et al. Phase I trial of the proteasome inhibitor PS-341 in patients with refractory hematologic malignancies. J. Clin. Oncol. 2002, 20, 4420‒4427. doi: 10.1200/JCO.2002.01.133
- Chauhan, D.; Singh, A.; Brahmandam, M.; et al. Combination of proteasome inhibitors bortezomib and NPI-0052 trigger in vivo synergistic cytotoxicity in multiple myeloma. Blood 2008, 111, 1654‒1664. doi: 10.1182/blood-2007-08-105601
- Pérez-Galán, P.; Roué G.; Villamor, N.; et al. The proteasome inhibitor bortezomib induces apoptosis in mantle-cell lymphoma through generation of ROS and noxa activation independent of p53 status. Blood 2006, 107, 257‒264. doi: 10.1182/blood-2005-05-2091
- Strauss, S.J.; Higginbottom, K.; Jüliger, S.; et al. The proteasome inhibitor bortezomib acts independently of p53 and induces cell death via apoptosis and mitotic catastrophe in B-cell lymphoma cell lines. Cancer Res. 2007, 67, 2783‒2790. doi: 10.1158/0008-5472.CAN-06-3254
- Hideshima, T.; Richardson, P.; Chauhan, D.; et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001, 61, 3071‒3076.
- LeBlanc, R.; Catley, L.P.; Hideshima, T.; et al. Proteasome inhibitor PS-341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. Cancer Res. 2002, 62, 4996‒5000.
- Cowan, A.J.; Green, D.J.; Kwok, M.; et al. Diagnosis and management of multiple myeloma: a review. JAMA 2022, 327, 464‒477. doi: 10.1001/jama.2022.0003
- Durie, B.G.M.; Hoering, A.; Abidi, M.H.; et al. Bortezomib with lenalidomide and dexamethasone versus lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma without intent for immediate autologous stem-cell transplant (SWOG S0777): a randomised, open-label, phase 3 trial. Lancet. 2017, 389, 519‒527. doi: 10.1016/S0140-6736(16)31594-X
- Chang, J.E.; Li, H.; Smith, M.R.; et al. Phase 2 study of VcR-CVAD with maintenance rituximab for untreated mantle cell lymphoma: an eastern cooperative oncology group study (E1405). Blood 2014, 123, 1665‒1673. doi: 10.1182/blood-2013-08-523845
- Chang, J.E.; Carmichael, L.L.; Kim, K.; et al. VcR-CVAD induction chemotherapy followed by maintenance rituximab in mantle cell lymphoma: a wisconsin oncology network study. Br. J. Haematol. 2011, 155, 190‒197. doi: 10.1111/j.1365-2141.2011.08820.x
- Richardson, P.G.; Sonneveld, P.; Schuster, M.; et al. Extended follow-up of a phase 3 trial in relapsed multiple myeloma: final time-to-event results of the APEX trial. Blood 2007, 110, 3557‒3560. doi: 10.1182/blood-2006-08-036947
- Robak, T.; Jin, J.; Pylypenko, H.; et al. Frontline bortezomib, rituximab, cyclophosphamide, doxorubicin, and prednisone (VR-CAP) versus rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) in transplantation-ineligible patients with newly diagnosed mantle cell lymphoma: final overall survival results of a randomised, open-label, phase 3 study. Lancet Oncol. 2018, 19, 1449‒1458.
- Dimopoulos, M.A.; Goldschmidt, H.; Niesvizky, R.; et al. Carfilzomib or bortezomib in relapsed or refractory multiple myeloma (ENDEAVOR): an interim overall survival analysis of an open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 1327‒1337. doi: 10.1016/S1470-2045(17)30578-8
- Kuhn, D.J.; Chen, Q.; Voorhees, P.M.; et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 2007, 110, 3281‒3290. doi: 10.1182/blood-2007-01-065888
- Wang, H.; Guan, F.; Chen, D.; et al. An analysis of the safety profile of proteasome inhibitors for treating various cancers. Expert Opin. Drug Saf. 2014, 13, 1043‒1054. doi: 10.1517/14740338.2014.939953
- Siegel, D.S.; Martin, T.; Wang, M.; et al. A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood 2012, 120, 2817‒2825. doi: 10.1182/blood-2012-05-425934
- Vij, R.; Wang, M.; Kaufman, J.L.; et al. An open-label, single-arm, phase 2 (PX-171-004) study of single-agent carfilzomib in bortezomib-naive patients with relapsed and/or refractory multiple myeloma. Blood 2012, 119, 5661‒5670. doi: 10.1182/blood-2012-03-414359
- Stewart, A.K.; Rajkumar, S.V.; Dimopoulos, M.A.; et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2015, 372, 142‒152. doi: 10.1056/NEJMoa1411321
- Vesole, D.H.; Bilotti, E.; Richter, J.R.; et al. Phase I study of carfilzomib, lenalidomide, vorinostat, and dexamethasone in patients with relapsed and/or refractory multiple myeloma. Br. J. Haematol. 2015, 171, 52‒59. doi: 10.1111/bjh.13517
- Lendvai, N.; Hilden, P.; Devlin, S.; et al. A phase 2 single-center study of carfilzomib 56 mg/m2 with or without low-dose dexamethasone in relapsed multiple myeloma. Blood 2014, 124, 899‒906. doi: 10.1182/blood-2014-02-556308
- Papadopoulos, K.P.; Siegel, D.S.; Vesole, D.H.; et al. Phase I study of 30-minute infusion of carfilzomib as single agent or in combination with low-dose dexamethasone in patients with relapsed and/or refractory multiple myeloma. J. Clin. Oncol. 2015, 33, 732‒739. doi: 10.1200/JCO.2013.52.3522
- Korde, N.; Roschewski, M.; Zingone, A.; et al. Treatment with carfilzomib-lenalidomide-dexamethasone with lenalidomide extension in patients with smoldering or newly diagnosed multiple myeloma. JAMA Oncol. 2015, 1, 746‒754.
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; et al. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017, 36, 561‒584. doi: 10.1007/s10555-017-9707-8
- Kupperman, E.; Lee, E.C.; Cao, Y.; et al. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010, 70, 1970‒1980. doi: 10.1158/0008-5472.CAN-09-2766
- Liu, R.; Fu, C.; Sun, J.; et al. A new perspective for osteosarcoma therapy: proteasome inhibition by MLN9708/2238 successfully induces apoptosis and cell cycle arrest and attenuates the invasion ability of osteosarcoma cells in vitro. Cell. Physiol. Biochem. 2017, 41, 451‒465. doi: 10.1159/000456598
- Chauhan, D.; Tian, Z.; Zhou, B.; et al. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin. Cancer Res. 2011, 17, 5311‒5321. doi: 10.1158/1078-0432.CCR-11-0476
- Richardson, P.G.; Kumar, S.K.; Masszi, T.; et al. Final overall survival analysis of the TOURMALINE-MM1 phase III trial of ixazomib, lenalidomide, and dexamethasone in patients with relapsed or refractory multiple myeloma. J. Clin. Oncol. 2021, 39, 2430‒2442. doi: 10.1200/JCO.21.00972
- Kale, A.J.; Moore, B.S. Molecular mechanisms of acquired proteasome inhibitor resistance. J. Med. Chem. 2012, 55, 10317‒10327. doi: 10.1021/jm300434z
- Chauhan, D.; Singh, A.V.; Ciccarelli, B.; et al. Combination of novel proteasome inhibitor NPI-0052 and lenalidomide trigger in vitro and in vivo synergistic cytotoxicity in multiple myeloma. Blood 2010, 115, 834‒845. doi: 10.1182/blood-2009-03-213009
- Singh, A.V.; Palladino, M.A.; Lloyd, G.K.; et al. Pharmacodynamic and efficacy studies of the novel proteasome inhibitor NPI-0052 (marizomib) in a human plasmacytoma xenograft murine model. Br. J. Haematol. 2010, 149, 550‒559. doi: 10.1111/j.1365-2141.2010.08144.x
- Spencer, A.; Harrison, S.; Zonder, J.; et al. A phase 1 clinical trial evaluating marizomib, pomalidomide and low-dose dexamethasone in relapsed and refractory multiple myeloma (NPI-0052-107): final study results. Br. J. Haematol. 2018, 180, 41‒51. doi: 10.1111/bjh.14987
- Hari, P.; Matous, J.V.; Voorhees, P.M.; et al. Oprozomib in patients with newly diagnosed multiple myeloma. Blood Cancer, J. 2019, 9, 66. doi: 10.1038/s41408-019-0232-6
- Shah, J.; Usmani, S.; Stadtmauer, E.A.; et al. Oprozomib, pomalidomide, and dexamethasone in patients with relapsed and/or refractory multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2019, 19, 570‒578. doi: 10.1016/j.clml.2019.05.017
- Sanderson, M.P.; Friese-Hamim, M.; Walter-Bausch, G.; et al. M3258 Is a selective inhibitor of the immunoproteasome subunit LMP7 (beta5i) delivering efficacy in multiple myeloma models. Mol. Cancer Ther. 2021, 20, 1378‒1387. doi: 10.1158/1535-7163.MCT-21-0005
- Manton, C.A.; Johnson, B.; Singh, M.; et al. Induction of cell death by the novel proteasome inhibitor marizomib in glioblastoma in vitro and in vivo. Sci. Rep. 2016, 6, 18953. doi: 10.1038/srep18953
- Rentsch, A.; Landsberg, D.; Brodmann, T.; et al. Synthesis and pharmacology of proteasome inhibitors. Angew. Chem., Int. Ed. Engl. 2013, 52, 5450‒5488. doi: 10.1002/anie.201207900
- Manach, C.; Scalbert, A.; Morand, C.; et al. Polyphenols: food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727‒747. doi: 10.1093/ajcn/79.5.727
- Smith, D.M.; Wang, Z.; Kazi, A.; et al. Synthetic analogs of green tea polyphenols as proteasome inhibitors. Mol Med. 2002, 8, 382‒392. doi: 10.1007/BF03402019
- Smith, D.M.; Daniel, K.G.; Wang, Z.; et al. Docking studies and model development of tea polyphenol proteasome inhibitors: applications to rational drug design. Proteins 2003, 54, 58‒70. doi: 10.1002/prot.10504
- Nam, S.; Smith, D.M.; Dou, Q.P. Ester bond-containing tea polyphenols potently inhibit proteasome activity in vitro and in vivo. J. Biol. Chem. 2001, 276, 13322‒13330. doi: 10.1074/jbc.M004209200
- Chen, D.; Landis-Piwowar, K.R.; Chen, M.S.; et al. Inhibition of proteasome activity by the dietary flavonoid apigenin is associated with growth inhibition in cultured breast cancer cells and xenografts. Breast Cancer Res. 2007, 9, R80. doi: 10.1186/bcr1797
- Singh, V.; Sharma, V.; Verma, V.; et al. Apigenin manipulates the ubiquitin-proteasome system to rescue estrogen receptor-β from degradation and induce apoptosis in prostate cancer cells. Eur. J. Nutr. 2014, 54, 1255‒1267. doi: 10.1007/s00394-014-0803-z
- Liu, F.T.; Agrawal, S.G.; Movasaghi, Z.; et al. Dietary flavonoids inhibit the anticancer effects of the proteasome inhibitor bortezomib. Blood 2008, 112, 3835‒3846. doi: 10.1182/blood-2008-04-150227
- Tsalikis, J.; Abdel-Nour, M.; Farahvash, A.; et al. Isoginkgetin, a natural biflavonoid proteasome inhibitor, sensitizes cancer cells to apoptosis via disruption of lysosomal homeostasis and impaired protein clearance. Mol. Cell. Biol. 2019, 9, e00489‒e00418. doi: 10.1128/MCB.00489-18
- Yin, R.; Li, T.; Tian, J.X.; et al. Ursolic acid, a potential anticancer compound for breast cancer therapy. Crit. Rev. Food Sci. Nutr. 2018, 58, 568‒574. doi: 10.1080/10408398.2016.1203755
- Li, S.; Kuo, H.D.; Yin, R.; et al. Epigenetics/epigenomics of triterpenoids in cancer prevention and in health. Biochem. Pharmacol. 2020, 175, 113890. doi: 10.1016/j.bcp.2020.113890
- Ahmad, M.F. Ganoderma lucidum:a rational pharmacological approach to surmount cancer. J. Ethnopharmacol. 2020, 260, 113047. doi: 10.1016/j.jep.2020.113047
- Dai, Y.; Desano, J.; Tang, W.; et al. Natural proteasome inhibitor celastrol suppresses androgen-independent prostate cancer progression by modulating apoptotic proteins and NF-kappaB. PLoS One 2010, 5, e14153. doi: 10.1371/journal.pone.0014153
- Walcott, S.E.; Heikkila, J.J. Celastrol can inhibit proteasome activity and upregulate the expression of heat shock protein genes, hsp30 and hsp70, in Xenopus laevis A6 cells. Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol. 2010, 156, 285‒293. doi: 10.1016/j.cbpa.2010.02.015
- Wang, W.B.; Feng, L.X.; Yue, Q.X.; et al. Paraptosis accompanied by autophagy and apoptosis was induced by celastrol, a natural compound with influence on proteasome, ER stress and Hsp90. J. Cell. Physiol. 2012, 227, 2196‒2206. doi: 10.1002/jcp.22956
- Sethi, G.; Ahn, K.S.; Pandey, M.K.; et al. Celastrol, a novel triterpene, potentiates TNF-induced apoptosis and suppresses invasion of tumor cells by inhibiting NF-κB-regulated gene products and TAK1-mediated NF-κB activation. Blood. 2006, 109, 2727‒2735. doi: 10.1182/blood-2006-10-050807
- Yang, H.; Shi, G.; Dou, Q.P. The tumor proteasome is a primary target for the natural anticancer compound withaferin A isolated from “Indian Winter Cherry”. Mol. Pharmacol. 2007, 71, 426‒437. doi: 10.1124/mol.106.030015
- Vanden Berghe, W.; Sabbe, L.; Kaileh, M.; et al. Molecular insight in the multifunctional activities of withaferin, A. Biochem. Pharmacol. 2012, 84, 1282‒1291. doi: 10.1016/j.bcp.2012.08.027
- Vanden Berghe, W.; Sabbe, L.; Kaileh, M.; et al. Development of withaferin A analogs as probes of angiogenesis. Bioorg. Med. Chem. Lett. 2006, 16, 2603‒2607. doi: 10.1016/j.bmcl.2006.02.039
- Tsukamoto, S.; Tane, K.; Ohta, T.; et al. Four new bioactive pyrrole-derived alkaloids from the marine sponge axinella brevistyla. J. Nat. Prod. 2001, 64, 1576‒1578. doi: 10.1021/np010280b
- Tsukamoto, S.; Tatsuno, M.; van Soest, R.W.; et al. New polyhydroxy sterols: proteasome inhibitors from a marine sponge acanthodendrilla sp. J. Nat. Prod. 2003, 66, 1181‒1185. doi: 10.1021/np030120v
- Tsukamoto, S.; Yamanokuchi, R.; Yoshitomi, M.; et al. Aaptamine, an alkaloid from the sponge aaptos suberitoides, functions as a proteasome inhibitor. Bioorg. Med. Chem. Lett. 2010, 20, 3341‒3343. doi: 10.1016/j.bmcl.2010.04.029
- da Silva, D.C.; Andrade, P.B.; Valentão, P.; et al. Neurotoxicity of the steroidal alkaloids tomatine and tomatidine is RIP1 kinase- and caspase-independent and involves the eIF2α branch of the endoplasmic reticulum. J. Steroid Biochem. Mol. Biol. 2017, 171, 178‒186. doi: 10.1016/j.jsbmb.2017.03.009
- Mohamed, I.E.; Kehraus, S.; Krick, A., et al. Mode of action of epoxyphomalins A and B and characterization of related metabolites from the marine-derived fungus paraconiothyrium sp. J. Nat. Prod. 2010, 73, 2053‒2056. doi: 10.1021/np100310k
- Goel, U.; Usmani. S.; Kumar, S. Current approaches to management of newly diagnosed multiple myeloma. Am. J. Hematol. 2022, 97(S1), S3‒S25. doi: 10.1002/ajh.26512
- Zhang, L.; Wu, M.; Su, R., et al. The efficacy and mechanism of proteasome inhibitors in solid tumor treatment. Recent Pat. Anticancer Drug Discov. 2022, 17, 268‒283. doi: 10.2174/1574892816666211202154536
- Roeten, M.S.F.; Cloos, J.; Jansen, G. Positioning of proteasome inhibitors in therapy of solid malignancies. Cancer Chemother. Pharmacol. 2017, 81, 227‒243. doi: 10.1007/s00280-017-3489-0
- Li, K.; Crews, C.M. PROTACs: past, present and future. Chem. Soc. Rev. 2022, 51, 5214‒5236. doi: 10.1039/D2CS00193D


